Salud
Oncología
:: PATOLOGÍA NEOPLÁSICA UROLÓGICA ::
-----------------------------------------
A un paciente de 45 años asintomático se le encuentra un nódulo renal de 2,5 cm. localizado en la corteza en un reconocimiento de salud. Se le interviene quirúrgicamente y se le reseca con un margen de tejido renal normal.
1.- Liste y describa los diferentes sustratos morfológicos de las masas renales de pequeño y gran tamaño localizadas en la corteza.
POR: HPB
TUMORES DEL RIÑÓN
En el riñon se producen tumores benignos y malignos . En general, los benignos son hallazgos casuales de autopsia y rara vez tienen importancia clínica. Por otra parte, los tumores malignos son muy importantes en la clínica y merecen un considerable interés. Con diferencia, el más frecuente de ellos es el carcinoma de células renales, seguido del tumor de Wilms, que aparece en los niños y, en tercer lugar, de los tumores uroteliales de los cálices y de la pelvis.
TUMORES BENIGNOS
ADENOMA CORTICAL: Los adenomas pequeños, discretos, se originan en los túbulos renales y son hallazgos de autopsia relativamente frecuentes (7al 22 %).
MORFOLOGIA. Estos tumores suelen medir menos de 2 cm de diámetro. Se encuentran siempre en el interior, de la corteza y consisten nódulos pequeños, de color gris amarillento, pálidos y aparentemente encapsulados ..Microscópicamente están formados por estructuras complejas. ramificadas y papilomatosas con numerosas vellosidades que se proyectan hacia un espacio quístico. Sus células .pueden diferenciarse a túbulos. Glándulas o cordones o crecer como masas totalmente indiferendadas.l tipo celular de todos estos patrones histológicos es bastante uniforme y no muestra atipias. Se trata de células de forma cúbica o poligonal con núcleos centrales pequenos y regulares y citoplasmas, que a veces se encuentran llenos de vacuolas lipidicas.
Con criterios meramente histológicos. no es posible diferenciar estos tumores del adenocarcinoma de células renales. Por ello. su tamaño se utiliza en el diagnostico diferencial. pues se admite que cuando son mayores de 3 cm de diámetro muestra tendencia a metastatizar mientras que los diámetros inferiores rara vez lo hacen. Este criterio sólo resulta útil, evidentemente, como regla general. Ya que los adenomas pueden dar lugar a adenocarcinoma. Ademas, aunque casi todos los adenomas renales son hallazgos causales de autopsia, algunos de tamaño limítrofe (2 a 3 cm) pueden ser identificados durante una exploración radiológica o una Intervención quirúrgica. Se aconseja considerar como, carcinomas precoces a todos estos tumores de tamaño intermedio.
FIBROMA O HAMARTOMA RENAL (Tumor renomedular de células intersticiales). Ocasionalmente, se encuentran en la autopsia pequeños focos de tejido de color blanco grisáceo y consistencia firme, por lo general de diámetro inferior a 1 cm, en el interior de las pirámides renales. El estudio microscópico revela células de aspecto fibroblástico y tejido colagenizado. Ultraestructuralmente, las células muestran las características de las célula intersticial renal. Estos tumores no tienen tendendencia a la transformación maligna.
ANGIOMIOLlPOMA. Se trata de un tumor benigno formado por vasos, músculo liso y tejido adiposo. Se encuentran angiomiolipomas en el 25 al 50 % de los pacientes con esclerosis tuberosa, enfermedad caracterizada por lesiones de la corteza cerebral que producen epilepsia y retraso mental, así como distintas anomalías cutáneas.
ONCOCITOMA. El oncocitoma es un tumor epitelial formado por grandes células eosinófilas de núcleo pequeño, redondeado, de aspecto benigno. El estudio ultraestructural demuestra que estas células contienen numerosas mitocondrias grandes. Macroscópicamente, estos tumores muestran un color pardo y un aspecto relativamente homogéneo encontrándose, por 10 general, bien encapsulados. Sin embargo, pueden adquirir un gran tamaño (hasta 12 cm de diámetro). Se han clasificado histológicamente en tres grados: los tumores de grado 1, que constituyen la mayoría, son siempre benignos y nunca metastatizan; los de grados más altos metastatizan en un pequeño porcentaje de los casos.
TUMORES MALIGNOS
Carcinoma de células renales ( Hipernefrona , adenocarcinoma renal )
Los carcinomas de células renales constituyen entre el 1 y el 3 % de todos los cánceres viscerales y justifican el 85 a 90 % de todos los cánceres renales del adulto. Son más frecuentes en las personas de edad avanzada, habitualmente en el sexto o séptimo decenios de la vida, y más frecuentes en el varón, con una relación entre sexos de 3:1. Su color amarillo y el parecido entre las células tumorales y las células claras de la corteza suprarrenal han hecho que también se les conozca como hipernefromas. En la actualidad, sin embargo, se sabe que surgen del epitelio tubular y, por tanto, se les denomina adenocarcinomas renales.
Los estudios epidemiológicos demuestran mayor incidencia de estas neoplasias en los fumadores de cigarrillos, pipa o cigarros puros. También intervienen factores genéticos . Así, casi las dos terceras partes de todos los pacientes con síndrome de Von Hippel-Lindau (VHL), caracterizado por la existencia de hemangioblastomas del sistema nervioso central y de la retina desarrollan carcinomas de células renales bilaterales y a menudo múltiples. EI gen supresor del crecimiento tumoral del VHL se encuentra en el brazo corto del cromosoma 3 (3p25-26) y codifica una proteína que participa en la transducción de las señales o en la adhesión celular72. En los casos familiares de carcinoma no papilar de células renales también se encuentran a menudo translocaciones 3:8 y 3:11, mientras que en casos esporádicos se han descrito mutaciones y deleciones. Así pues, la investigación actual implica al gen VHL o a un gen relacionado con el VHL, situado en el cromosoma 3, en la carcinogénesis renal. Los carcinomas papilares muestran otras alteraciones cromosómicas.
MORFOLOGIA. Este tumor presenta un aspecto macroscópico muy característico. Puede aparecer en cualquer parte del riñon, pero es más frecuente en los polos, sobre todo en el superior. En general. Se trata, de lesiones unilaterales solitarias con forma de masa esférica y diámetros que oscilan entre 3y15cm. Estan formados por un tejido de color amarillo , gris y blanco brillante que distorsiona la arquitectura renal Presentan grandes áreas de necrosis Isquémiças opacas y grisáceas, focos de hemorragia y zonas de reblandecimiento. Sus bordes suelen esta bien delimitados por la cápsula renal.
Sin embargo en el parénquima circundante poden, encontrarse pequeños nódulos satélites, como demostración evidente de la agresividad de es tumores, A medida que crece, puede hacer prominencia ,hacia los cálices, la pelvis y atravesar , en ultima instancia, las paredes del sistema colector para' Infiltrar Incluso el uréter. Una de sus caracteristicas más llamativas es su capacidad para invadir la vena renal y crecer formando una sólida columna en su interior. Su crecimiento continuado alcanza la vena cava e Incluso las cavidades derechas del corazón. Histológicamente. Los patrones de crecimiento varían desde el papilar al sólido, trabecular (en cordones) o tubular (como túbulos). En cualquier tumor pueden encontrarse, mezclas variables de todos los patrones de diferenciación. EI tipo celular más frecuente (70 %) es el constituido por células claras, de contorno, redondeado o poligonal y citoplasma claro y abundante. Con técnicas especiales, se demuestra que este último contiene glucógeno y lípidos., EI .15 % de, estos 'tumores 'son papilares y están, formados, por células claras ô células granulosas (carcinoma renal de células granulosas), de citoplasma eosinófilo granuloso que crece con un patrón sarcomatoide y 'tiene un pronóstico claramente más desfavorable. Casi todos los carcinomas están bien diferenciados (grados I y II ) , pero algunos. (grado IV) muestran grandes 'atípias nucleares, núcleos abigarrados y células gigantes: su estroma suele ser escaso pero muy vascularizado.
EVOLUCIÓN CLÍNICA. Las tres características diagnósticas clásicas, dolor costo vertebral, masa palpable y hematuria, sólo aparecen, por desgracia, en el 10 % de los casos. De las tres, la más fiable es la hematuria, que se produce en uno u otro momento en alrededor del 90 % de los pacientes. Sin embargo, esta hematuria suele ser intermitente y puede ser microscópica, por lo que el tumor sólo se diagnostica cuando alcanza un gran tamaño. En ese momento, suele haber dado ya lugar a síntomas generales como fiebre, malestar general, debilidad y pérdida de peso. Este patrón de crecimiento asintomático es muy frecuente, de modo que la neoplasia puede haber alcanzado más de 10 cm de diámetro en el momento del diagnóstico.El carcinoma de células renales es considerado como uno de los grandes «imitadores» de la medicina, pues tiende a producir distintos síntomas sistémicos no relacionados con el riñón. Además de la fiebre y otros síntomas generales, pueden dar lugar a varios síndromes paraneoplásicos debidos a la producción anormal de hormonas, tales como policitemia, hipercalcemia, hipertensión, disfunción hepática, feminización o masculinización, síndrome de Cushing , eosinofilia, reacciones leucemoides y amiloidosis. Una de sus características más frecuentes es su tendencia a metastatizar ampliamente antes de producir síntomas o signos locales. En el 25 % de los pacientes con carcinoma de células renales de nuevo diagnóstico se encuentran signos radiográficos de metástasis en el momento de su presentación. La diseminación del tumor afecta sobre todo los pulmones (más del 50 %) y los huesos (33 %); seguidos, por orden descendente de frecuencia, de los ganglios linfáticos regionales, el hígado, las suprarrenales y el encéfalo. En el 10 al 15 % de los casos, el tumor primario cruza la línea media y metastatiza en el riñón contralateral. Un diagnóstico 10 más precoz posible es fundamental para el tratamiento de los carcinomas de células renales. Este diagnóstico se hace, habitualmente, durante la investigación de una hematuria en un paciente de edad madura o anciano. La ecografia renal, Ia nefrotomografia y Ia pielografia permiten hacer el diagnóstico diferencial entre esta neoplasia y los quistes simples. La citología urinaria también puede ser útil, cuando demuestra la presencia de células tumorales en la orina. La supervivencia media los 5 anos de los pacientes con carcinoma de células renales es de alrededor del 45 %, ascendiendo hasta el 70 % en ausencia de metástasis a distancia. Cuando existen
invasión de la vena renal o extensión a la grasa perirrenal, la cifra cae al 15-20 % . La nefrectomía es el tratamiento de elección.
:: Bibliografía ::
- Patologia Estructural y Funcional, 5ª ed. Aut: Stanley l-Robbins editorial Mc Gran-Hill interamericana ed 1995
- WebPathology.com, Dharam M. Rammani MD
- Anatomia Patológica. Autor Dra Juliana Fariñas Editora Salvat Editores S.A. Edición 1990
- Universidad de Campinas, www.fcm.unicamp.com.br/departamentos/anatomia.
- Cátedra de Anatomia Patológica , Hospital de Clínicas ,2004 Dra ML Musto
- Liang Cheng, MD, Dept. of Pathology, Indiana University School of Medicine, Indianapolis.
:: PATOLOGÍA NEOPLÁSICA UROLÓGICA ::
-----------------------------------------
A un paciente de 45 años asintomático se le encuentra un nódulo renal de 2,5 cm. localizado en la corteza en un reconocimiento de salud. Se le interviene quirúrgicamente y se le reseca con un margen de tejido renal normal.
2.- Señale cuales con los factores que debe estudiar y referir en el informe anatomopatológico el patólogo con vistas a establecer un pronóstico.
POR: Daniel Quirós Zamorano
En este supuesto clínico se hace referencia a que tras la intervención quirúrgica en un paciente previamente asintomático (se descubre la lesión durante una revisión) se reseca una masa o nódulo de 2,5 cm. con márgenes de tejido renal normal. Para introducir como se realizaría el estudio anatomopatológico de dicho nódulo y el subsiguiente establecimiento del pronóstico haremos una serie de consideraciones previas muy generales referidas a las masas renales.
El tumor o masa renal es un área anormal dentro del riñón. Los términos masa, lesión y tumor se intercambian con frecuencia. Los tumores pueden ser benignos o malignos. El tipo más común de tumor renal es un área llena de líquido llamada quiste. El quiste simple es benigno y tiene una apariencia típica en los estudios de imagen. Este quiste no progresa hacia cáncer y, por lo general, no requiere de tratamiento de seguimiento. El quiste complejo no tiene la apariencia típica del benigno y puede contener en si mismo una lesión cancerosa. Cuando se presenta el quiste complejo, las necesidades individuales determinarán la necesidad del tratamiento. Otro tipo de tumor renal es un tumor sólido de riñón (es decir, sin líquido). Los tumores renales sólidos pueden ser benignos, pero generalmente son malignos; de hecho, más del 90% de los tumores renales sólidos lo son. Dentro de éstos, la gran mayoría son adenocarcinomas con todas sus variantes (células claras, cromófobo…), con lo que nuestro trabajo se centrará fundamentalmente en los datos histopatológicos que estudiaremos en el caso de los adenocarcinomas renales o carcinoma de células renales (CCR).
Macroscópicamente, estos tumores renales son grandes masas, generalmente lobuladas, que distorsionan el riñón y que afectan la grasa perirrenal. Suelen estar cubiertos por una pseudocápsula fibrosa, por efecto de los tejidos comprimidos por el tumor y el estroma reaccional (no es una verdadera cápsula), lo que les da un aspecto globuloso. Al corte, puede aparecer de color gris amarillento con zonas quísticas hemorrágicas y de consistencia variable, desde zonas duras a zonas blandas, afectadas por la necrosis. También pueden presentar calcificaciones. La fascia de Gerota limita la extensión fuera del riñón. Las estructuras vecinas pueden ser invadidas en las grandes masas tumorales.
Tradicionalmente se consideraba que cualquier masa renal de menos de 3 cm. se correspondía con un adenoma y mayor de ese tamaño sería un carcinoma. Actualmente este concepto está totalmente desechado por incierto, entre otros motivos porque los adenomas a pesar de su pequeño tamaño son entidades muy discutidas, consideradas por algunos autores como lesión premaligna y para muchos otros como una lesión cancerosa propiamente dicha, algo que se apoya en el hecho de que diferenciar un adenoma de un adenocarcinoma bien diferenciado microscópica e histológicamente puede ser tremendamente complicado (el adenoma suele tener una arquitectura histológica túbulo papilar y con frecuencia no tiene cápsula). Además existen carcinomas de menos de 3 cm. con todas las características de malignidad, con lo cual el tamaño no permite discriminar entre benignos y malignos.
En el caso que nos ocupa, pese a tener 2,5 cm., al ser una lesión única y de consistencia sólida podría tratarse perfectamente de un CCR, los cuales suelen presentarse de forma unilateral, afectando a parte o a todo el riñón, siendo un 90% de todas las masas sólidas renales. Tras la resección de la pieza el anatomopatólogo deberá estudiar los siguientes factores histopatológicos que son los que nos permitirán establecer un pronóstico:
-
Estadio Patológico.
-
Grado Nuclear de Fuhrman.
-
Tipo Histológico.
-
Invasión Microvascular.
-
Presencia o Ausencia de Cambio Sarcomatoide.
-
Márgenes Quirúrgicos.
-
Otros.
ESTADIO PATOLÓGICO.
El estadio patológico se puede determinar por distintos sistemas, aunque todos presentan equivalencias entre si:
-
pTNM
-
Estadiaje de la AJCC
-
Estadiaje de Robson
pTNM
La clasificación pTNM para el estadio patológico en tumores renales se resume en la siguiente tabla:
| Tumor Primario (pT) | |
| pTx | El tumor primario no puede ser medido. |
| pT0 | No existe evidencia de tumor primario. |
| pT1 | Tumor de 7 cm. o menor en su diámetro mayor, limitado al riñón. |
| PT1a | Tumor de 4 cm. o menor en su diámetro mayor, limitado al riñón. |
| PT1b | Tumor de más de 4 cm. pero no mayor de 7 cm. en su diámetro mayor, limitado al riñón. |
| pT2 | Tumor mayor de 7 cm. en su diámetro mayor, limitado al riñón. |
| pT3 | Tumor que se extienda a los vasos principales o invade la glándula adrenal o tejidos perirrenales, pero no traspasa la fascia de Gerota |
| pT3a | Tumor que invade la glándula adrenal o tejido perirrenal, pero no traspasa la fascia de Gerota |
| pT3b | Tumor que se extiende groseramente a la vena renal o la vena cava. |
| pT3c | Tumor que se extiende groseramente sobre la vena cava bajo el diafragma. |
| pT4 | Tumor que traspasa la fascia de Gerota. |
| Ganglios Linfáticos regionales (pN) | |
| pNx | Los ganglios regionales no pueden ser valorados. |
| pN0 | No existe afectación ganglionar regional. |
| pN1 | Metástasis en un único ganglio, de 2 cm. o menos de diámetro mayor. |
| pN2 | Metástasis en un único ganglio, de más de 2 cm. pero no mayor de 5 cm. de diámetro mayor, o múltiples ganglios linfáticos, ninguno mayor de 5 cm. de diámetro máximo. |
| pN3 | Metástasis en un ganglio linfático de más de 5 cm. de diámetro. |
| Nota: la contralateralidad no afecta la clasificación. | |
| Metástasis a Distancia (pM) | |
| pMx | La presencia de metástasis no puede ser evaluada. |
| pM0 | No existen metástasis a distancia. |
| pM1 | Existen metástasis a distancia. |
El estadio patológico siempre se consideró el factor más importante para predecir la supervivencia de pacientes con CCR. Los tumores confinados a un órgano tienen buen pronóstico, con una probabilidad de supervivencia específica al cáncer a los 5 años desde 70% hasta 90%. La estadificación pTNM divide los CCR localizados en dos categorías: T1 y T2 (masas pequeñas y grandes, 1974; < 2.5 cm y > 2.5 cm, 1987; < 7 cm y > 7 cm, 1997). La versión de pTNM del 2002 agregó una subestadificación de los tumores T1 (< 7 cm) en T1a y T1b, con un corte entre ellos en los 4 cm. El fundamento de esta nueva clasificación fue identificar los candidatos a cirugía conservadora de la nefrona. Lo anterior fue apoyado por un estudio de Hafez y col. y por los resultados prometedores en la cirugía conservadora de la nefrona.
Muchos artículos no apoyan esta división y proponen diferentes cortes entre los estadios T1 y T2, la mayoría alrededor de los 5 cm. Ficarra y col., en una serie bicéntrica, demostraron no sólo que los 5.5 cm son el corte ideal, sino que los 7 cm no tienen significado pronóstico según el análisis multivariable. Como comentó Novick, estos datos proveen un importante motivo para una redefinición del actual sistema pTNM. Las neoplasias con afectación de la cápsula renal (que afectan la grasa perirrenal o la glándula suprarrenal) tienen peor pronóstico, con una supervivencia a los 5 y 10 años de 57% y 35%, respectivamente. Algunos autores sugieren que los tumores que invaden la grasa renal podrían tener un comportamiento más agresivo por su más fácil acceso al sistema vascular. La afectación adrenal ipsilateral se clasifica como estadio T3a, lo mismo que la invasión a la grasa perirrenal. Puede apreciarse en 1% a 5% de los pacientes con CCR y puede asociarse con pronóstico aún peor que el considerado hasta el momento. Sandock y col. informaron que 5.3% de los pacientes con afectación adrenal ipsilateral fallecieron por progresión de la enfermedad en una mediana de tiempo de 7 meses. Sagalowsky y col. mostraron que 81% de los pacientes con afectación suprarrenal ipsilateral murieron por la enfermedad dentro de los 26 meses. Las recomendaciones pTNM son para clasificar los pacientes con afectación suprarrenal como T3a sólo si esto es expresión de una contigüidad local y no de diseminación de la enfermedad.
Un estudio reciente de la UCLA muestra peor pronóstico para los tumores con invasión directa de la glándula suprarrenal (0% de supervivencia específica al cáncer a 5 años), que para los tumores con invasión de la grasa perirrenal (36% de supervivencia específica al cáncer a 5 años). Esto sugiere que aquellos tumores que invaden la glándula suprarrenal deberían ser considerados como tumores T4. Los datos sugieren realizar adrenalectomía ipsilateral sólo en presencia de neoplasmas en el polo superior o grandes masas con afectación adrenal radiológicamente comprobada.
El CCR se extiende hacia la vena renal en 10% a 20% de los casos y hacia la vena cava inferior (T3b, T3c) en 4% a 10% de los casos. Es esperable que estas presentaciones predigan un resultado oncológico muy desfavorable. Sin embargo, series publicadas informan tasas de supervivencia específicas al cáncer a los 5 años de 47% a 68% si el trombo renal es removido completamente. En varias series, el nivel del trombo (vena renal, vena cava o aurícula derecha) no parece afectar el pronóstico. Parecen ser más importantes como factores pronósticos la invasión de grasa perirrenal, el estado del nodo linfático o la invasión de la pared venosa por el trombo. Ficarra y col. informaron que la implicancia del eje venoso es insignificante si no está asociada con factores pronósticos desfavorables. Se demostró que la enfermedad más allá de la fascia de Gerota (T4) y la enfermedad nodal empeoran el pronóstico. La supervivencia a los 5 años de las dos categorías es de 16% a 28% para la enfermedad T4, y de 8% a 35% en el compromiso nodal. El papel de la linfadenectomía aún no se determinó. Algunos autores mencionan tasas de supervivencia a los 5 años de 50% en enfermedad nodal tratada con linfadenectomía extensa, mientras otros adjudican a la linfadenectomía una real ventaja en el pronóstico de 4% a 6%.
La enfermedad metastásica tiene pronóstico muy malo, con supervivencia a los 5 y 10 años de 5% a 10% y de 0 a 7% respectivamente. La única opción terapéutica para estos pacientes es la combinación de nefrectomía citorreductiva radical e inmunoterapia (IL-2 o interferón), con una respuesta completa informada de 3% a 43% y respuesta parcial de 10% a 91%.
Los ensayos aleatorizados EORTC y SWONG mostraron demora en la progresión de la enfermedad (5 meses contra 3 meses) y mejor supervivencia (11 a 17 meses contra 7 a 8 meses) en pacientes con enfermedad metastásica tratada con terapias combinadas en relación con los tratados sólo con inmunoterapia.
Estadiaje de la AJCC
La clasificación AJCC (American Joint Committee on Cancer) es paralela a la pTNM y se pueden establecer equivalencias entre una y otra. Consta de cuatro estadios:
-
Estadio I: T1, N0, M0
-
Estadio II: T2, N0, M0
-
Estadio III: T1-2, N1, M0; o bien T3a-c, N0-1, M0
-
Estadio IV: T4; o bien cualquier T, N2, M0; o bien cualquier T, Cualquier N, M1.
Estadiaje de Robson.
Se trata de un estadiaje que se basa en la modificación del sistema de Flocks y Kadesky, siendo realmente sencillo de aplicar, y muy utilizado en la clínica. Robson utilizó esta clasificación para correlacionar el estadio en la presentación con el pronóstico. Así se obtuvieron 4 estadios: :
-
Estadio I: Tumor confinado al riñón. Se corresponde con los estadios I y II de la AJCC (T1-2, N0)
-
Estadio II: Afectación de los tejidos perirrenal y/o adrenales, pero todavía dentro de la fascia de Gerota. Se corresponde con el estadio III de la AJCC (T3a, N0).
-
Estadio III: Invasión regional con afectación de la vena renal o de la vena cava inferior, adenopatías regionales, o ambas. Se corresponde con el estadio de la AJCC: T3b, N0, M0; o bien cualquier T, N1-3, M0
-
Estadio IV: Enfermedad avanzada con extensión directa a órganos adyacentes, o con metástasis a distancia. Corresponde con estadio de la AJCC: T4, cualquier N, M0; o bien cualquier T, cualquier N, M1.
GRADO NUCLEAR DE FUHRMAN
Existen varios sistemas de gradación nuclear, pero ninguno de ellos está libre de problemas para ser reproducido o respecto de la variabilidad entre observadores.
El sistema de grados nucleares de Fuhrman es el más usado en Europa y América del Norte. Es un sistema de cuatro niveles, basado en la morfología del núcleo y en la presencia del nucleolo. Varios artículos notifican la utilidad de este sistema para predecir la supervivencia específica al cáncer independientemente del estadio patológico. Su principal problema es su escasa capacidad para identificar diferentes resultados en las distintas categorías. Los cuatro niveles o grados nucleares de Fuhrman son los siguientes:
-
Grado 1 (G1): Núcleo redondeado, uniforme, 10 µm. de diámetro, nucleolo inconspicuo o ausente.
-
Grado 2 (G2): Núcleo levemente irregular, 15 µm. de diámetro, nucleolo evidente.
-
Grado 3 (G3): Núcleo muy irregular, 20 µm., nucleolo grande y prominente.
-
Grado 4 (G4): Núcleos bizarros y multilobulados, 20 µm., nucleolo prominente, cromatina en grumos.
En relación con las series de Fuhrman, la mayoría de los artículos publicados muestran una diferencia en la supervivencia específica al cáncer sólo confrontando algunas categorías, en particular tumores G1-2 contra G3-4, G1 contra G2 contra G3-G4, o G1-G2 contra G3 contra G4.
Esta variabilidad podría deberse a una moderada concordancia entre observadores, y a la migración de grado imputable a la fijación tisular subóptima. Bretheau y col. informaron alta coincidencia entre diferentes patólogos en lo referido a grados nucleares. Todas las otras series publicadas no confirmaron estos datos, mencionan un acuerdo moderado entre observadores en sistemas de dos y cuatro niveles.
El papel del tipo histológico no está aún bien establecido. Los diferentes tipos pueden tener distintos pronósticos. Además, es difícil la comparación de grados entre los diferentes tipos histológicos. La UICC y la AJCC recomiendan asignar el grado nuclear sólo a los CCR convencional y papilar. Desgraciadamente, la mayoría de los artículos publicados no subdividen sus pacientes en diferentes tipos histológicos.
TIPO HISTOLÓGICO.
La clasificación histológica más usada para el CCR es la de Heidelberg, que distingue entre carcinoma de células claras convencional (75% a 80%), papilar (10% a 15%), cromofóbo (5%), de conductos de Bellini (< 1%) y los no clasificados (5%). El impacto pronóstico de los diferentes tipos histológicos es todavía controversial y difícil de demostrar. Gran parte de los casos corresponden a series anteriores que deberían ser reclasificadas.
Bonsib y col. mostraron tasas de supervivencia a los 5 años de 55% a 60% para el CCR claras convencional, de 80% a 90% para el papilar y de 90% para el cromófobo. Moch y col., en una serie de 588 pacientes, no encontraron ninguna diferencia en el pronóstico entre el papilar y el convencional. Cheville y col., por el contrario, en una serie mayor (2 385 pacientes), tras elanálisis univariado mostraron tasas de supervivencia de 10 años de diferencia entre el CCR convencional contra el papilar y el cromófobo. Los tumores de los conductos colectores de Bellini son raros pero muy agresivos, con 67% de los casos fallecidos a los 2 años del diagnóstico, y una mediana de supervivencia de 9 meses.
INVASIÓN MICROVASCULAR.
Para la detección de la existencia de invasión microvascular se inspecciona a alto aumento (X200 o superior) la periferia del tumor. Se considera que existe invasión microvascular cuando encontramos un grupo de células tumorales rodeadas de células endoteliales que han sido marcadas positivamente con anticuerpos frente antígenos del Factor VIII y/o del CD34.
Se correlaciona con el tamaño del tumor, el grado de diferenciación y el estadío tumoral. Ningún paciente en estadío I tiene invasión microvascular, estando presente en el 50 % de los estadío III y IV. Sólo el 6 % de los pacientes sin invasión microvascular progresan, en comparación con el 39 % que la presentan.
PRESENCIA O AUSENCIA DE CAMBIO SARCOMATOIDE.
La diferenciación o cambio sarcomatoide, alguna vez reconocida como un subtipo diferente, se considera, de acuerdo con la clasificación de Heidelberg, como un componente que puede estar asociado a todos los tipos histológicos de CCR. Tiene impacto pronóstico negativo debido, entre otras cosas, a su comportamiento agresivo, con una supervivencia media de 9 meses y presentación metastásica en 77% de los casos.
Macroscópicamente se trata de lesiones blancas, mal definidas, con necrosis y/o hemorragias. Al microscopio se observan células neoplásicas atípicas, fusiformes, de aspecto sarcomatoso, CK positivas.
MÁRGENES QUIRÚRGICOS.
Una vez resecada la lesión, generalmente con márgenes de tejido sano al sospecharse que se trata de un tumor maligno, siempre deben inspeccionarse dichos márgenes, pues aunque desde el punto de vista macroscópico parezcan limpios, microscópicamente pueden verse afectados, lo cual modifica el pronóstico y la consiguiente actuación terapéutica.
Así los bordes de la lesión pueden no estar invadidos por células carcinomatosas, o bien es posible que si exista dicha invasión, y en este segundo caso puede llegar a afectar a: la cápsula renal, grasa perirrenal, vena renal, fascia de Gerota, Ureter, etc... según el tamaño y localización de la neoplasia. De existir invasión se debe completar la resección del riñón, bien aumentando los márgenes de la misma, o bien realizando una nefrectomía total.
OTROS.
Existen otras técnicas que nos van a ayudar en el estudio anatomopatológico de la pieza, aunque tienen una menor relevancia que los anteriores. Entre ellos podemos destacar:
-
Ploidía del ADN
Di Silverio y col. informaron diferentes progresiones de tumores localizados de acuerdo con la ploidía del ADN y estratificaron los datos para diferentes tamaños de tumor. Abou-Rebyeh y col. hallaron diferentes riesgos de progresión, de acuerdo con la ploidía tumoral, y estratificaron los datos por estadio del tumor, con diferencias extraordinarias (6% contra 92% para T1-T2 contra 100% para T3).
-
Morfometría Nuclear
La morfometría nuclear fue propuesta como factor pronóstico por Carducci y col., quienes informaron correlación entre el espectro de elipticidad y la supervivencia sin recurrencia tras el análisis multivariado. Otras series encontraron correlación entre otros parámetros morfométricos y la supervivencia sin recurrencia, pero esta información no fue significativa en análisis multivariados.
-
Marcadores Moleculares
Varios marcadores moleculares fueron señalados como predictores del pronóstico en el CCR. Desgraciadamente, la investigación aún no demostró un nivel de precisión lo suficientemente alto. El grupo de trabajo Nº 4 de la UICC y la AJCC asignaron la categoría III a la mayoría de los marcadores moleculares. Por ejemplo, la proliferación (fracción de fase S, PCNA), marcadores de apoptosis (p53, bcl-2, p21), factores de crecimiento, moléculas de adhesión celular, angiogénesis, factores de respuesta del huésped, genes de supresión tumoral, factores de resistencia, citoquinas y anormalidades citogenéticas. Los únicos marcadores moleculares que pertenecen a la categoría II son AgNORs y Ki67. Estudios recientes apoyan su valor pronóstico en análisis multivariables.
-
Promedio de Mitosis
Su número es bajo en los carcinomas renales de bajo grado. Su número es alto en los carcinomas renales agresivos. La supervivencia a 10 años es de 67 y 16 %, respectivamente.
CONCLUSIÓN
Los tumores sólidos de riñón más frecuentes son los Carcinomas de Células Renales, con todas sus variantes, siendo un 90% del total, y de entre ellos cabe destacar el Carcinoma renal de células claras con más de un 80% de los casos de tumores malignos. El principal factor pronóstico para la supervivencia global es el estadio tumoral, aunque para el cáncer de riñón el valor del tamaño tumoral puede tener menos importancia. En general, el pronóstico de los pacientes con este tipo de lesiones empeora a medida que aumenta el tamaño tumoral y con la existencia de metástasis linfáticas regionales y con la existencia de metástasis a distancia.
Los tumores confinados al riñón (T1 y T2) tienen muy buen pronóstico, con supervivencias a los 5 años en torno al 92% - 95%. E los casos de afectación extrarrenal, hacia la vena cava o los ganglios locales, la supervivencia a los 5 años no supera el 40% - 50%, aunque parece tener un pronóstico significativamente mejor la extensión a la vena cava que la afectación ganglionar. La supervivencia a los 5 años si hay afectación metastásica es muy pobre, sin superar el 0% - 20%. Los resultados de supervivencia a los 5 años por estadios serían:
-
Estadio I: 90 - 100 %
-
Estadio II: 75 - 95 %
-
Estadio III: 60 - 70 %
-
Estadio IV: 15 - 30 %
También son muy importantes en el pronóstico el grado nuclear de las células que forman parte de dicho tumor, siendo de peor pronóstico un mayor grado nuclear de Fuhrman. Éste está estrechamente relacionado con el tipo histológico de los CCR, siendo mayor la supervivencia a los 5 años en la variante cromófoba, hasta un 90%. Debemos señalar por último que tienen un valor pronóstico negativo: la presencia de invasión microvascular, que se de cambio sarcomatoide, y que exista afectación de los márgenes quirúrgicos de la resección.
TRATAMIENTO
De una manera muy breve y para que este trabajo no quede incompleto procederemos a describir el tratamiento básico de los CCR, que como ya se ha comentado en varias ocasiones, son con diferencia el tumor sólido de riñón más frecuente.
Este tratamiento puede ser un tanto diferente dependiendo del tamaño, zona afectada y características especiales del tumor:
-
Carcinoma renal localizado: nefrectomía radical o parcial si es monorrenal anatómico o funcional, cáncer bilateral, enfermedad de von Hippel Lindau, tumor único menor de 4 cm., etc.
-
Carcinoma renal con trombo tumoral: abordaje de acuerdo a las características del trombo.
-
Carcinoma extendido a estructuras vecinas: cirugía para mejorar la calidad de vida del paciente.
-
Tratamiento de las metástasis: si es única valorar su resección, si son múltiples: citoquina, inmunoterapia, terapia génica, vacunas, implante de células stem, tratamientos multimodales.
Las respuestas frente a los distintos tipos de tratamiento son:
-
Interferón alfa: 10% respuestas parciales, 2% respuestas completas.
-
Interleukina II: 16% respuesta, 20% de supervivencia a 5 años y 10% a los 10.
-
Interleukina II + interferón alfa + 5 fluorouracilo: 32% de respuesta.
-
Tratamiento multimodal: cirugía + tratamientos biológicos o viceversa
Por último en el carcinoma renal metastásico, en pacientes en buenas condiciones generales, la resección del tumor primario se realiza para mejorar la calidad de vida del paciente, pudiendo haber mejoría del estado general. Si hay compromiso de los órganos adyacentes, la resección debe realizarse más para mejorar la calidad de vida que la supervivencia. El rol de la nefrectomía en la enfermedad metastásica renal debe ser selectivo, siendo necesario evaluar riesgos y beneficios respecto de la calidad de vida del paciente y de su supervivencia.
:: Bibliografía ::
- How is Kidney cancer (Renal Cell carcinoma) Staged?. American Cancer Society, Dirección web: http://www.cancer.org/docroot/CRI/content/CRI_2_4_3X_How_is_kidney_cancer_staged_22.asp
- Factores Pronósticos en el Carcinoma de Células Renales. Vicenzo Ficarra et al. Departament of Urology. University of Verona. Publicado para SIIC.
- Factores Pronósticos en los Tumores Urológicos. M.T. Dueñas, et al. Servicio de Oncología Radioterápica, Hospital General Yagüe, Burgos. Anales del sistema sanitario de Navara. Dirección Web: http://www.cfnavarra.es/salud/anales/textos/vol24/suple1/suple12.html
- Significance of Angiogenesis and Microvascular Invasion in Renal Cell Carcinoma. Yoram DEKEL, et al. Departments of 1Urology and 2Pathology, Hasharon Hospital, Rabin Medical Center, Campus Golda, Petah Tikva, and Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel. PATHOLOGY ONCOLOGY RESEARCH, Vol 8, nº 2, 2002.
- Kidney. Protocol applies to all invasive carcinomas of renal tubular origin. It excludes Wilms tumors and tumors of urothelial origin. John R. Srigley,et al. Department of Laboratory Medicine, Credit Valley Hospital, Mississauga, Ontario, Canada. Protocol revision date: January 2005. Based on AJCC/UICC TNM, 6th edition.
- Información General sobre el Cáncer Renal. Cancer Consultants, oncology resource center. Dirección web: http://patient.cancerconsultants.com/treatment.aspx?lang=en&id=914
- Tumores de Riñón. Primeras Jornadas Urológicas de San Julián. Sociedad Urológica de la Patagonia. 30 de Noviembre de 2001.
- Tumores Renales. Dra. ML Musto, Hospital de Clínicas, Cátedra de Anatomía Patológica, CICLIPA I. 2004.
- Principios de Práctica de Oncología. Tomo I, páginas: 839-849. 2ª Edición. Vicent T. et al. Salvat Editores 1988.
- Oncología Médica. Tomo I, páginas: 699-714. H. Cortés Funes. et al. Ed. Nova Sidonia - Oncología Grupo Aula Médica. 1999.
- Patología Quirurgica. Tomo III, páginas: 1587-1599. J.L. Balibrea Cantero. et al. Marbán Libros 2002.
:: PATOLOGÍA NEOPLÁSICA UROLÓGICA ::
-----------------------------------------
Un niño de 2 años presenta una masa sólida de 8 cm localizada en el riñón derecho.
1.- Liste y describa los diferentes sustratos morfológicos de las masas renales en el niño.
POR: Marcelo Kluge
:: TUMORES RENALES PEDIÁTRICOS ::
- Tumores nefroblásticos:
-
Nefroblastoma (TW ): de histología favorable o con anaplasia ( difusa o focal)
-
Restos nefrogénicos y nefroblastomatosis
-
Nefroma quístico
-
Nefroblastoma quístico, parcialmente diferenciado
-
Tumores metanéfricos y entidades relacionadas :
-
Adenoma metanéfrico
-
Adenofibroma metanéfrico
-
Tumor estromal metanéfrico
- Nefroma mesoblástico: celular, clásico y mixto
- Sarcoma de células claras
- Tumor rabdoide
- Tumores epiteliales renales de la infancia:
-
Carcinoma papilar
-
Carcinoma medular
-
Tumores renales asociados a la t Xp11.2
- Tumores raros :
-
Tumor osificante renal de la infancia
-
Angiomiolipoma
Dado que el 80% corresponde al TW y es muy baja la incidencia del resto de los
tumores nombrados, es básico conocerlo bien como punto de partida de una búsqueda bien orientada de diagnóstico diferencial.
Nefroblastoma (TW)
Representa el 6% de los tumores pediátricos con una incidencia anual de 650 casos nuevos en EEUU . La edad promedio al diagnóstico es de 36 a 42 meses, 98% antes de los 10 años. Presentación clínica: masa abdominal, dolor, vómitos, anorexia, hipertensión arterial (25%) y anomalías congénitas asociadas. El TW típicamente recapitula el desarrollo normal del riñón a partir del metanefros y los genes WT1 Y Pax 2 inducen en este proceso, entre otros eventos, la conversión de mesénquima en epitelio. En el riñón normal en desarrollo se expresa WT1 en el blastema metanéfrico y en el epitelio glomerular, persistiendo en el adulto sólo su expresión en podocitos. Mutaciones del WT1 como la deleción del cromosoma 11 en la banda 11p13 se ha asociado al TW pero sólo se halla presente en el 5-20% de los casos esporádicos, < del 10% de los constitucionales y < del 30% de los bilaterales.
Síndromes genéticos y genes implicados:
Locus Gen
WARG 11p13 WT1
Daniss. Drash 11p13 WT1
Beckwith-Wiedemann 11p15 WT2
Li-Fraumeni 17p13 p53
TW familiar 17q12-21 FTW1
17q13.3-13.4 FWT2
Por otra parte la mutación del gen p53 se asocia a anaplasia y se registraron cambios secunadarios en los cromosomas 1p16q.
Macroscopía: masa única, redondeada,bien delimitada del parénquima adyacente , con cápsula fibrosa peritumoral. Tamaño variable (peso promedio 550gr). Superficie de corte pálida mucoide, uniforme y consistencia blanda, puede estar septado con aspecto lobular, 5 % multicéntrico y puede raramente presentarse como masa botrioide en pelvis. En ocasiones hay formas quísticas, multiloculadas. El protocolo de examen macroscópico debe incluir: localización, tamaño, color, consistencia, grado de necrosis, evaluación del seno renal, cápsula tumoral, cápsula renal, vena renal, grasa perirrenal, uréter y ganglios regionales.
Histología: La mayoría presenta tres componentes: blastema metanéfrico, epitelio y estroma ( formas trifásicas) que representan cada uno y en su conjunto, estadíos de la nefrogénesis, pudiendo ser también bi o monofásicos.
Componente blastemal: células pequeñas, redondas o poligonales, estrechamente agrupadas, con mínimo citoplasma, núcleos regulares con cromatina granular y pequeño nucléolo no siempre visible, con numerosas mitosis. Pueden adoptar un patrón nodular, basaloide o serpenteado. Cuando su distribución es difusa pierde la cohesividad celular y presenta mayor agresividad invasiva local y vascular.
Componente epitelial: túbulos, papilas, patrón rosetoide o formación de estructuras glomeruloides que carecen en general de capilares. Pueden mostrar distintos estadíos madurativos e incluso diferenciación epitelial heteróloga con epitelio mucinoso, ciliado o pavimentoso.
:: Tubular ::
:: Glomerular ::
Componente estromal : Células fusiformes con fondo mixoide que remeda al mesénquima embrionario presente en casi todos los casos , fibroblastos y músculo liso con distintos grados de diferenciación y elementos mioblásticos primitivos similares a los de la capa de cambium del rabdomiosarcoma botrioide. Dentro de los tejido heterólogos puede verse: cartílago, osteoide, tejido adiposo, células ganglionares maduras., tejido neuroglial, y destacamos especialmente al músculo esquelético por ser el más frecuente, definiendo incluso una variante rabdomiomatosa fetal de nefroblastoma.
:: O componente blastematoso é o predominante. Forma ilhotas de células pouco diferenciadas, às vezes com necrose central ::
Todo lo descrito hasta aquí corresponde a los TW con histología favorable.
Un 5 % de casos son de histología desfavorable que se define por la presencia de Anaplasia cuyos criterios diagnósticos son:
-
agrandamiento nuclear ( 3 veces el tamaño del núcleo blastemal) con
hipercromasia.
-
mitosis poliploides multipolares.
Estos casos son edad dependiente (menores de 2 años), más frecuentes en raza negra, y de peor pronóstico, no por mayor agresividad sino por mayor resistencia a la quimioterapia.
Por otra parte tiene implicancia pronóstica si la anaplasia es focal o difusa.
Anaplasia Focal:
1) 1 o pocos focos de anaplasia bien definidos, rodeados por tumor no anaplásico
2) anaplasia confinada al parénquima renal sin compromiso vascular
3) ausencia de severo pleomorfismo nuclear e hipercromasia en tumor no
anaplásico.
La presencia de anaplasia en una biopsia pretumorectomía o prenefrectomía es indicador de anaplasia difusa.
Nefroma mesoblástico congénito (NMC)
El NMC es el tumor renal más frecuente en menores de 3 meses y el 98% ocurre antes del año de vida. Clínica :masa abdominal, polihidramnios, hidrops, hipercalcemia ( tumor productor de prostaglandina E).
Macroscopía: masa solitaria, unilateral de 1 a 14 cm, sólida y firme indistinguible del TW.Puede haber necrosis y hemorragia. Se localiza cercano al hilio y compromete al seno renal.
Histología: neoplasia monomorfa fusocelular compuesta por células de línea fibroblástica o miofibroblástica. Se describen 3 patrones: clásico, celular y mixto.
NMC clásico: remeda a la fibromatosis infantil. Fascículos de células ahusadas con escasas fibras colágenas, espacios vasculares dilatados de paredes finas y actividad mitótica variable. Se extiende al parénquima adyacente y tejidos perirrenales. Presencia de elementos renales atrapados, cambios metaplásicos con aspecto embrionario y nódulos de cartílago hialino (cambios displásicos). No tiene diferenciación de músculo esquelético.
NMC celular: representa un fibrosarcoma infantil intrarrenal. Alta densidad celular con aspecto sarcomatoso, células de núcleos vesiculares con leve a moderado pleomorfismo y escaso citoplasma. A diferencia del anterior es bien circunscripto. La vascularización capilar prominente es semejante al sarcoma de células claras.
Sarcoma de células claras ( SCC)
Mayor incidencia en el segundo año de vida. Sexo masc./ fem. = 2/1. Los raros casos de adultos son idénticos a los pediátricos. Da metástasis óseas. Se caracteriza por una proliferación de células indiferenciadas, uniformes, con abundante matriz extracelular, separadas por una fina trama vascular y células ahusadas,en cordones y nidos.
Aunque macroscopicamente tenga un aspecto bien delimitado, en la histología es característica la infiltración celular del parénquima adyacente, que produce atropamiento de estructuras renales normales en la periferia, rasgo similar al visto en el NMC y en el tumor rabdoide, no así en el TW. Presenta diferentes patrones histológicos que dificultan su diagnóstico ya que mimetiza al resto de tumores renales: mixoide, esclerosante, celular, epitelioide, fusocelular, en empalizada, pericitomatoso. El 3% presenta franca anaplasia.
Tumor Rabdoide (TR)
Representa sólo el 2,5% de los tumores renales pediátricos pero es el de mayor mortalidad.Mayor incidencia a los 11 meses (menores de 3 años). Sexo masc./fem.=1,5/1. El 15% se asocia a tumores de SNC.
Se caracteriza por una población monomorfa de células grandes, no cohesivas, con bordes bien delimitados, núcleo vesiculoso y nucléolo prominente, citoplasma con ocasionales inclusiones acidofílicas. Se dispone en playas.Es altamente infiltrativo local y vascular. Patrones: esclerosante, epitelioide, ahusado y linfomatoide.
Carcinoma de células renales pediátrico
Son muy poco frecuentes. Edad 9-10 años. Suelen asociarse al síndrome Von Hippel-Lindau lo que sugiere ciertas diferencias con los carcinomas renales de adultos, y poseen cambios genéticos distintivos. Incluyen: carcinoma de células renales papilar (CRP) , carcinoma medular y carcinoma de células renales con traslocación Xp11.2.
CRP: es el más común de los tres, y plantea el diagnóstico diferencial con el TW epitelial y el adenoma metanéfrico (AM).
| Pseudocápsula | Mitosis | Células | IHQ | |
| CRP | Prominente | Ocasionales | Columnares ,nucléolo prominente | EMA y CQ 7+ |
| TW epit. | “ | “ | Columnares, cromatina fina | WT1+ CQ+ focal |
| AM | Ausente | Ausentes | Ovoides, pequeño nucléolo | EMA- , CQ7+/-focal |
Tumores Metanéfricos:
Espectro de lesiones benignas que derivan del blastema metanéfrico.
Componente epitelial:
-
Adenoma metanéfrico
-
Tubular, glomeruloide, o papilar
-
Ausencia de mitosis y de nucléolo prominente
-
Ausencia de compromiso vascular
-
Negatividad con EMA y neg. variable con citoqueratina 7
-
Positividad con WT1, Cd56 y CD57.
Componente estromal:
-
Tumor estromal metanéfrico
-
Aspecto nodular con bajo aumento
-
Collaretes concéntricos rodeando túbulos y/o vasos
-
Angiodisplasia
-
Elementos heterólogos; cartílago, glia, grasa.
-
Positividad con CD34
Tumores renales pediátricos simil carcinoma renal de células claras
-
Angiomiolipoma epitelioide
-
Carcinoma renal t (Xp11)
-
Sarcoma alveolar de partes blandas
-
Tumor cromófobo u oncocítico
:: Bibliografía ::
- Trabajo Tumores Renales Pediátricos. Dra.Elena De Matteo , Hospital de Niños Ricardo Gutiérrez, Bs.As.
- Tumors of the kidney, bladder, and relates urinary structures, Murphy W, et al. AFIP, serie 4, 2004 : 8-99.
- Recent advances in pediatric renal neoplasia, Argani P,Ladanyi M,Adv.Anat Pathol 10, (5) 2003: 243-260.
- Renal neoplasia: WHO 2003.The increasing influence of molecular pathology.
- Rrevised SIOP Working Classification of renal tumors of childhood, Vujanic G, et al, Med Pediatr Oncol 2002;38:77-82.
- Management of WILM's Tumour: current practice and future goals, Coppes M et al, Lancet Oncol 2004;5: 37-46.
- Protocol for the examination of specimens from patient with Wilms Tumor or other renal tumors of childhood. Qualman S et al. Arch Pathol Lab Med 2003;
vol 127 : 1280-1289.
- Myogenesis in Wilm's tumors is associated with mutations of the WT1 gene and activation of Bcl-2 and the Wnt signaling pathway. Fukuzawa R, et al. Pediatric and developmental pathology, 2004; 7: 125-137.
- Metanephric stromal tumor with urothelial extension. Lorenzo a. et al. J of
Urology 2003;169 : 1095-1097.
- Metanephric adenoma, nephrogenic rests and Wilm's tumor: a histologic and immunophenotypic comparison.Lager DJ et al. Am J Surg Pathol 2001;25 (10):
1290-6.
- ETV6 rearrangements in patient with infantile fibrosacomas and congenital mesoblastic nephromas by fluorescence in situ hybridization. Adem C, et al. Mod Pathol 2001; 14(12): 1246-51.
- Renal cell carcinoma in children: a clinicopathologic study. Indolfi P, et al. J of Clinical Oncology 2003 ,21(3): 530-535.
- Neuroblastoma mimicking Rhabdoid tumor of the kidney. Shaw P,Dickman P. J of Pediatric Hematol Oncology, 2003;25 (7): 572-574.
:: PATOLOGÍA NEOPLÁSICA UROLÓGICA ::
-----------------------------------------
Un niño de 2 años presenta una masa sólida de 8 cm localizada en el riñón derecho.
2.- Señale cuales son los factores que debe estudiar y referir en el informe anatomopatológico el patólogo con vistas a establecer un pronóstico.
POR: José Santiago Alonso Mate
Tumor de Wilms y otros tumores renales infantiles
-
Información general
-
Tumor de Wilms
-
Sarcoma de células claras
-
Tumores rabdoides del riñón
-
Tumores neuroepiteliales del riñón
-
Nefroblastoma cístico parcialmente diferenciado
-
Clasificación celular
-
Tumor de Wilms
-
Estadio I (43% de los pacientes)
-
Estadio II (23% de los pacientes)
-
Estadio III (23% de los pacientes)
-
Estadio IV (10% de los pacientes)
-
Estadio V (5% de los pacientes)
-
Anaplasia estadio I-IV
-
Información sobre los estadios
-
Tumor de Wilms
-
Sarcoma de células claras del riñón
-
Tumor rabdoide del riñón
-
Aspectos generales de las opciones de tratamiento
-
Tumor de Wilms en estadio I
-
Tumor de Wilms en estadio II
-
Tumor de Wilms en estadio III
-
Tumor de Wilms en estadio IV
-
Tumor de Wilms en estadio V
Tumor de Wilms:
Es una enfermedad curable en la mayoría de los niños afectados. En los Estados Unidos se diagnostican más de 500 casos cada año. Más del 90% sobreviven 4 años después del diagnóstico, lo cual constituye un avance si se compara con el 80% de supervivencia que se observaba entre el 1975 y 1984. El pronóstico está relacionado no sólo con el estadio de la enfermedad en el momento del diagnóstico, las características histopatológicas del tumor, la edad del paciente y el tamaño del tumor, sino también con la estrategia en equipo para cada paciente por parte del cirujano pediatra, el radiooncólogo y el oncólogo pediatra. Ensayos clínicos anteriores han evaluado en parte, con cierto grado de éxito, si una terapia reducida es suficiente para controlar la enfermedad en pacientes en estadio inicial con tumor de Wilms de histología favorable.
Clasificación celular
Tumor de Wilms
Aunque la mayoría de los pacientes con un diagnóstico histológico del tumor de Wilms responden bien a los tratamientos actuales, aproximadamente 10% de los pacientes tienen características histopatológicas que están relacionadas con un pronóstico más precario y en algunos tipos, con una alta incidencia de recidiva y muerte. El tumor de Wilms puede dividirse en dos grupos pronósticos sobre la base de su histopatología:
-
Histología favorable: Histológicamente imita el desarrollo del riñón normal y consiste en tres tipos de células: blastémicas, epiteliales (túbulos) y estromátícas. No todos los tumores son trifásicos, y los patrones monofásicos pudieran presentar dificultades al diagnóstico. No existe anaplasia en el tumor.
-
Histología anaplástica: Puede ser focal o difusa (pleomorfismo y atipia celulares extremos). La anaplasia focal no confiere un pronóstico precario, mientras que la anaplasia difusa sí (excepto en el estadio I). La anaplasia está relacionada con la resistencia a la quimioterapia y podría ser detectada después de aplicar quimioterapia preoperatoria.
Según la Clasificación Histológica (SIOP 2001), esta tiene presente tres grupos de riesgo atendiendo a los diferentes componentes titulares, su maduración y distinta arquitectura, definiendo así el t. de Wilms trifásico mixto, de predominio epitelial y/o estromal, y contemplando la presencia o no de anaplasia focal o difusa, además de tener presente el pretratamiento oncológico por el que se añade, el tipo necrótico y el blastematosos. A tener en cuenta es, quepara la denominación de cada tipo, es necesario contener 2/3 del tejido predominante, y nunca más del 10% de blastema. El de tipo blastematoso tendrá igualmente 2/3 del mismo. El subtipo rabdiomiomatoso estromal no muestra diferentes criterios, al igual que el nefroblastoma extrarrenal.
Es importante por otro lado definir la anaplasia difusa, con presencia de mitosis multipolares, núcleos x 3, y con hipercromasia. Añadiremos como signos relevantes a tener en cuenta, que, es el único tumor renal que es bilateral o multifocal, que tiene restos nefrogénicos entre el 25 y 40%, que contiene tejido adiposo y muscular estriado, así como túmulos neoplásicos.
-
Se puede decir que dos grandes grupos de investigadores abanderan el estudio del tumor de Wilms: la Société du Oncologie Pédiatrique (SIOP) y el Nacional Wilms Tumor Study (NWTS); ambos grupos han definido sistemas de clasificación histológica cuya correspondencia puede verse reflejada en la tabla posterior. La comparación entre ambos sistemas debe hacerse considerando el hecho de que la mayoría de los pacientes integrados en el protocolo SIOP reciben quimioterapia previa al examen patológico, mientras que en el NWTS las piezas quirúrgicas son examinadas sin terapia previa; este hecho limita la comparación histopatológica y de estadiaje en ambos grupos. Sucede así en tumores de gran agresividad morfológica e intensa actividad proliferativa cuando son examinados sin tratamiento previo, pueden aparecer mayoritariamente necróticos si son examinados tras quimioterapia citorreductora; asimismo un tumor que parece infiltrar la cápsula renal antes de la terapia puede aparecer limitado al riñón después de ella.
-
Macroscópicamente el TW tiene una apariencia sólida y lobulada, de consistencia blanda. Se encuentra rodeado de una pseudocápsula constituida por tejido renal adyacente comprimido y atrófico. Al corte presenta un color gris pálido con áreas intercaladas de hemorragia y necrosis así como zonas quísticas que pueden ser predominantes. Al contrario que el neuroblastoma, la presencia de calcificaciones es muy infrecuente. Su friabilidad le hace sensible a la ruptura en el curso de una palpación abdominal especialmente traumática o durante el acto quirúrgico con riesgo de diseminación peritoneal.
-
Microscópicamente, el rasgo más característico en el TW, es su diversidad estructural. Derivado del primitivo blastema metanéfrico reproduce el desarrollo normal del riñon con sus tres componentes (blastema, epitelio y estroma) cuya representación varía ampliamente de un tumor a otro. Pueden coincidir los tres tipos histológicos (trifásicos), pero lo más habitual es que coexistan dos de ellos (difásicos y mixtos). En un amplio estudio del German Pediatric ONcology el subtipo más frecuente fue el mixto seguido por el de predominio blastematoso y en tercer lugar el epitelial; los TW de predominio estromal fueron claramente los más infrecuentes. Clínicamente los tumores de predoninio blastematoso se comportan agresivamente con mayor proporcion de estadios avanzados al diagnóstico y con mayor facilidad para la diseminación metastásica; el subtipo epitelial se caracteriza pos su marcada diferenciación hacia túbulos y glomérulos con menor agresividad clínica. Los tumores de predominio estromal exhiben un amplio rango de diferenciación mesenquimal que incluye músculo liso, estriado, grasa, cartílago y osteoide.
-
Sea cual sea el subtipo histológico, es de gran interés la presencio o no de anaplasia por su demostrada asociación con resistencia a la quimioterapia. Morfológicamente se define por células en las que el diámetro mayor de su núcleo es al menos tres veces superior al de las células vecinas con incremento en su contenido cromático y presencia de figuras mitóticas hiperdiploides. Un 5% del total de los TW muestan anaplasia, siendo extemadamente rara por debajo de los 2 años de edad e incrementando progresivamente su frecuencia hasta superara el 10% en los pacientes mayores de 5 años.
-
Otros hallazgos morfológicos de interés lo constituyen la existencia o no de asociación a lesiones potencialmente precursoras de TW conocidas como restos nefrogenéticos y nefroblastomatosis. Los restos nefrogenéticos se encuentran en el 1% de las autopsias realizadas en neonatos que han muerto en circunstancias diversas, de lo que se deduce que estas lesiones regresan espontáneamente en la mayoría de los casos. Estos restos pueden ser perilobares y menos frecuentemente intralobares, encontrándose una de las dos variantes hasta en el 25% de los riñones con TW.
-
La nefroblastomatosis consiste en la presencia de restos nefrogénicos de formamultifocal o difusa en uno o ambos riñones; su inciencia oscila entre el 25-40% de los riñones con TW y representa un riesgo importante de desarrollar un nuevo tumor en el tejido renal restante por lo que estos pacientes deben ser evaluados periódicamente.
Sarcoma de células claras
Los sarcomas de células claras del riñón (CCSK, por sus siglas en inglés) no es una variante de los tumores de Wilms, pero si un tumor renal primario importante relacionado de forma significativa con una tasa alta de recaída y muerte en comparación con los tumores de Wilms de histología favorable. Además de las metástasis pulmonares, los sarcomas de células claras se propagan al hueso, cerebro y tejidos blandos. El patrón clásico de CCSK se define por una red de células y cuerdas separadas por una septa fibrovascular espaciadas entre si de forma regular en arborización.
Tumores rabdoides del riñón
Inicialmente se pensaba que este era una variante de rabdomiosarcomatoide del tumor de Wilms, pero se trata de un tipo de tumor altamente maligno. Las características más distintivas de los tumores rabdoides del riñón son células bastante grandes con núcleos vesiculares grandes, un nucleolo prominente único y, en algunas células, la presencia de inclusiones citoplásmicas eosinófilas globulares. La célula de origen se desconoce. Una presentación clínica diferente con fiebre, hematuria, edad joven (promedio de 11 meses) y un estadio tumoral alta al momento de la presentación, indica un diagnóstico de tumor rabdoide del riñón (RTK, por sus siglas en inglés). El RTK tiende a hacer metástasis no solo a los pulmones, sino también al cerebro. Entre un 10 a un 15% de los pacientes con RTK, presentan también lesiones del sistema nervioso central.
La lesión molecular característica que encontramos en los tumores rabdoides del riñón es una mutación en la pérdida de funcionalidad del gen hSNF5/INI1 el cual está localizado en la banda cromosómica 22q11.2. Esta misma anomalía molecular la encontramos en los tumores del sistema nervioso central, conocidas con el término tumores atípicos teratoides y rabdoides sin complicación al riñón. Algunos pacientes con tumores rabdoides tienen mutaciones constitucionales del gen hSNF5/INI1, y estos niños están en riesgo creciente de desarrollar un segundo tumor cerebral primario.
Tumores neuroepiteliales del riñón
Los tumores primarios neuroepiteliales del riñón (NETK, por sus siglas en inglés) son extremadamente poco comunes y muestran una tendencia singular hacia los adultos jóvenes. Es un neoplasma altamente agresivo, el cual presenta más comúnmente penetración de la cápsula renal, extensión hacia la vena renal y metástasis. Los NETK consisten de tumores neuroectodermales primitivos (PNET, por sus siglas en inglés) caracterizados por CD99 (MIC-2) inmunotinción y el EWS/FLI-1 o productos de fusión genética o carcinoma de células pequeñas estrechamente relacionados caracterizados por cromogranina positiva. Los dos subtipos podrían resultar difíciles de distinguir. En ambos tipos de NETK, se han visto características histológicas focales, atípicas tales como sarcoma de células claras, tumor rabdoide, tumores malignos de la vaina del nervio periférico y paraganglioma.
Nefroblastoma cístico parcialmente diferenciado
El nefroblastoma cístico parcialmente diferenciado (CPDN, por sus siglas en inglés) es una variante cística poco común del tumor de Wilms (1%) con características patológicas y clínicas distintivas. Existe una variedad de características patológicas que distinguen este neoplasma del típico tumor de Wilms. Los pacientes con enfermedad en estadio I tienen un 100% de tasa de supervivencia con cirugía sola. Pacientes con enfermedad en estadio II tienen un excelente resultado con resección del tumor seguida de vincristina y dactinomicina postoperativa.
Información sobre los estadios
Tumor de Wilms
Podemos decir que el estadiaje, se va a realizar atendiendo al estudio macroscópico y microscópico, y que habitualmente será un grado por encima del que nos refiera el cirujano. Para ello se procederá a la llegada de la pieza, a valorar la superficie externa cápsular renal, el hilio con los vasos renales y ureter, el peso, fotografia, teñido con tinta china antes de su apertura. Una vez realizada ésta, mediremos el tumor, y tendremos en cuenta el sinus, la o las cápsulas tumorales y la interfase tumor/riñón, y tenerlo presente en el tallado. De las áreas tumorales más preservadas tomaremos muestras para congelar a -80 grados.
El estadio se determina de acuerdo con los resultados de los estudios imagenológicos, quirúrgicos y patológicos de la nefrectomía, y es el mismo para tumores con características histológicas favorables o anaplásicas. Por lo tanto, los pacientes deberán ser caracterizados utilizando ambos criterios (por ejemplo, estadio II, histología favorable o estadio II, histología anaplásica ).
El sistema de estadificación empleado por el National Wilms' Tumor Study Group) y la incidencia por estadio se exponen someramente más adelante.
Estadio I (43% de los pacientes)
Para clasificarse en el estadio I del tumor de Wilms, deben satisfacerse 1 o más de los criterios siguientes:
-
El tumor está limitado al riñón y se ha extirpado completamente.
-
La superficie de la cápsula renal está intacta.
-
El tumor no se rompe ni antes de la escisión ni durante la misma.
-
El tumor no se abre ni se le hace biopsia (abierta o de aguja) antes de resecarlo.
-
No hay complicación a los vasos sinusoidales renales.
-
No hay tumor residual evidente más allá de los márgenes de escisión.
Estadio II (23% de los pacientes)
Para clasificarse en el estadio II del tumor de Wilms, deben satisfacerse 1 o más de los criterios siguientes:
-
El tumor se extiende más allá del riñón pero se ha resecado completamente.
-
No hay tumor residual aparente en los márgenes de la escisión ni más allá de ellos.
También puede existir cualquiera de estas otras situaciones:
-
Complicación tumoral de los vasos sanguíneos del seno renal, fuera del parénquima renal o en ambos lugares.
-
Se ha realizado la biopsia del tumor antes de resecarlo o hay derrame accidental local de tumor durante la operación, limitado al costado.
-
Extensión regional del tumor, es decir, penetración a través de la superficie externa de la cápsula renal en el tejido blando perirrenal o más de 1 a 2 mm de invasión tumoral en seno renal.
-
Los vasos fuera del riñón están infiltrados o contiene trombo tumoral.
Estadio III (23% de los pacientes)
Para clasificarse en el estadio III del tumor de Wilms, deben satisfacerse 1 o más de los criterios siguientes:
-
Tumor primario irresecable, debido a la infiltración local en estructuras vitales.
-
Metástasis a ganglio linfático. Estos ganglios, pueden encontrarse en el hilio renal y en las cadenas periaorticas.
-
Márgenes quirúrgicos positivos. El tumor se extiende más allá de los márgenes quirúrgicos microscópicos o macroscópicos.
-
Derrame accidental del tumor que complica las superficies peritoneales, ya sea antes de la operación o después de ella, o transección de trombo tumoral.
Estadio IV (10% de los pacientes)
El tumor de Wilms en estadio IV se define como la presencia de metástasis hematógena (pulmonar, hepática, ósea o cerebral) o metástasis a ganglios linfáticos fuera de la región abdominopelviana.
Estadio V (5% de los pacientes)
El tumor de Wilms en estadio V se define como complicación renal bilateral en el momento del diagnóstico inicial.
En los pacientes comprometidos de forma bilateral, se deberá intentar clasificar cada lado según los criterios anteriores (estadios I a III) en base al grado de la enfermedad antes de la biopsia. La supervivencia a 4 años era del 94% para aquellos pacientes cuya lesión más avanzada estaba en estadio I o II; y de 76% para aquellos cuya lesión más avanzada estaba en estadio III.
Anaplasia estadio I-IV
Los niños con tumores anaplásticos en estadio I tienen un pronóstico excelente. En su manejo puede emplearse el mismo régimen que se les da a los pacientes en estadio I de histología favorable. Los niños con anaplasia difusa en estadio II hasta la estadio IV, sin embargo, representan un grupo de mayor riesgo. Estos tumores son mucho más resistentes a la quimioterapia que tradicionalmente se administra a niños con tumor de Wilms (con histología favorable).
Tumor de Wilms en estadio I
Independientemente de la histología, todos los pacientes con tumores de Wilms en estadio I tienen un pronóstico excelente con el mismo tratamiento.
Tumores de histología favorable (FH, por sus siglas en inglés) (92% 4 años de supervivencia sin recaídas [RFS, por sus siglas en inglés]; 98% supervivencia de 4 años):
-
Nefrectomía con muestras de ganglios linfáticos y 18 semanas de quimioterapia con vincristina y pulso intensivo de dactinomicina.
Anaplasia focal o difusa (87,5% 2 años RFS, 85,5% supervivencia de 2 años):
-
Nefrectomía con muestras de ganglios linfáticos irradiación abdominal y 18 semanas de quimioterapia con vincristina y pulso intensivo de dactinomicina.
Podría ser posible el tratar a un subconjunto de pacientes solamente con cirugía sin incluir quimioterapia. El Grupo Oncológico Infantil (COG, por sus siglas en inglés) está planificando llevar a cabo un estudio numeroso para dilucidar este aspecto. En el Grupo 5 del Estudio Nacional de Tumores de Wilms (NWTS-5, por sus siglas en inglés) se trató con cirugía sola a niños menores de 24 meses de edad con tumor de Wilms en estadio I/HF y cuyo espécimen obtenido mediante nefrectomía pesó <550 gramos. El estudio se proyecta con una estricta regla para terminarlo (análisis provisional de la supervivencia sin recaida [RFS, siglas en inglés] de aproximadamente 90%), que fue excedida, lo que exigia el cierre del estudio. Se pudo rescatar a la mayoria de los pacientes exitosamente, no obstante, con una tasa de supervivencia de 100% a los 2 años.
Tumor de Wilms en estadio II
Tumores de histología favorable (85% 4 años de supervivencia sin recaída; 96% de supervivencia a 4 años):
-
Nefrectomía con muestras de ganglios linfáticos y 18 semanas de quimioterapia con vincristina y pulso intensivo de dactinomicina.
Anaplasia focal:
-
Nefrectomía con muestras de ganglios linfáticos irradiación abdominal y 24 semanas de quimioterapia con vincristina, doxorubicina y pulso intensivo de dactinomicina.
Anaplasia difusa (70% 4 años de supervivencia):
-
Nefrectomía con muestras de ganglios linfáticos irradiación abdominal y 24 semanas de quimioterapia con vincristina, doxorubicina, etopósido, ciclofosfamida y mesna.
Tumor de Wilms en estadio III
Tumores de histología favorable (90% 4 años de supervivencia sin recaída; 95% 4 años de supervivencia) con anaplasia focal o sin ella:
-
Nefrectomía con muestras de ganglios linfáticos irradiación abdominal y 24 semanas de quimioterapia con vincristina, doxorubicina y pulso intensivo de dactinomicina.
Anaplasia difusa (56% 4 años de supervivencia):
-
Nefrectomía con muestras de ganglios linfáticos, irradiación abdominal y 24 semanas de quimioterapia con vincristina, doxorubicina, etopósido, ciclofosfamida y mesna.
Tumor de Wilms en estadio IV
Tumores de histología favorable (80% 4 años de supervivencia sin recaída; 90% 4 años de supervivencia):
-
Nefrectomía con muestra de ganglios linfáticos, irradiación abdominal de acuerdo al estadio local del tumor renal, irradiación pulmonar bilateral para los pacientes con radiografía torácica que muestre metástasis pulmonares, y 24 semanas de quimioterapia con vincristina, doxorubicina y pulso intensivo de dactinomicina.
Anaplasia focal
-
Nefrectomía con muestra de ganglios linfáticos, irradiación abdominal de acuerdo al estadio local del tumor renal, irradiación pulmonar bilateral para los pacientes con radiografía torácica que muestre metástasis pulmonares, y 24 semanas de quimioterapia con vincristina, doxorubicina y pulso intensivo de dactinomicina.
Anaplasia difusa (17% 4 años de supervivencia)
-
Nefrectomía con muestra de ganglios linfáticos, irradiación abdominal, irradiación de todo el pulmón para los pacientes con radiografía torácica que muestre metástasis pulmonares, y 24 semanas de quimioterapia con vincristina, doxorubicina, etopósido, ciclofosfamida y mesna.
Tumor de Wilms en estadio V
El tratamiento de los niños con tumor de Wilms bilateral debe individualizarse. La terapia tiene como objetivo de erradicar todo el tumor y de preservar la mayor cantidad de tejido renal normal posible, con esperanza de reducir el riesgo de insuficiencia renal crónica en estos niños. Estudios han demostrado que no hay diferencia en supervivencia entre niños a quienes se somete a una biopsia bilateral seguida de quimioterapia y luego una resección quirúrgica, comparados con aquellos pacientes a los que se sometió a resección primero seguida de quimioterapia. Al principio, el paciente debe someterse a biopsias renales bilaterales con estadificación de cada riñón. No debe intentarse la escisión del tumor primario, pero los pacientes deben recibir quimioterapia preoperatoria. El tratamiento inicial se hace según el régimen EE-4A si los tumores renales presentan histología favorable y no son más extensos que los de estadio II. Los pacientes con tumores en estadio más avanzado y enfermedad de histología favorable recibirían, además, doxorrubicina (DD4A); los de histología anaplásica también deben recibir ciclofosfamida (Régimen I). Después de 6 semanas de quimioterapia el paciente es revaluado. Si los estudios de imágenes en serie no muestran una reducción tumoral, se debe llevar a cabo una cirugía de segunda exploración (nefrectomía parcial o corte mediante incisión) si se pueden obtener márgenes negativos, de otro modo habría que hacer otra biopsia para confirmar los tumores viables. La quimioterapia, la radioterapia o ambas después de dicha operación depende de la respuesta a la terapia inicial, requiriéndose terapia más intensiva para los pacientes con respuesta inadecuada a la terapia inicial observada en el segundo procedimiento.
Aproximadamente el 10% de los pacientes con tumores bilaterales tienen histología anaplásica y pueden beneficiarse de quimioterapia y radioterapia más intensivas y un enfoque quirúrgico agresivo en la operación de segunda observación.
PRONÓSTICO
Como resumen en cuanto al Pronóstico, podemos decir que la tasa de curación es de aproximadamente el 50%. La tasa de supervivencia global es del 80% o más durante periodos superiores a 15 años de seguimiento. Se puede considerar como factores pronósticos principales, el estadio tumoral en el momento del diagnóstico y la subhistología. La afectación de los ganglios linfáticos, también se ha asociado a un peor pronóstico, la contaminación abdominal por rotura del tumor disminuyendo la supervivencia.
Factores pronósticos NWTS-3
Histología Estadio Supervivencia/4 años
FAVORABLE I 96.7%
II 92.3%
III 87.3%
IV 83.4%
DESFAVORABLE I-III 71.6%
IV 55.2%
El sarcoma de células claras, es un tumor de alta malignidad, que tiende a recidivar localmente, supervivencia del 75% a los 4 años. El tumor rabdoide del riñón tiene una supervivencia del 25% a los 4 años.
:: Bibliografía ::
- Zuppan CW, Beckwith JB, Luckey DW Anaplasia in unilateral Wilms' tumor: a report from the National Wilms' Tumor Study Pathology Center. Hum Pathol 19 (10): 1199-209, 1988
- Vujanić GM, Harms D, Sandstedt B, et al. New definitions of focal and diffuse anaplasia in Wilms tumor: the International Society of Paediatric Oncology (SIOP) experience. Med Pediatr Oncol 32 (5): 317-23, 1999.
- Faria P, Beckwith JB, Mishra K, et al. Focal versus diffuse anaplasia in Wilms tumor--new definitions with prognostic significance: a report from the National Wilms Tumor Study Group. Am J Surg Pathol 20 (8): 909-20, 1996.
- Argani P, Perlman EJ, Breslow NE, et al. Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms Tumor Study Group Pathology Center. Am J Surg Pathol 24 (1): 4-18, 2000.
- Amar AM, Tomlinson G, Green DM, et al. Clinical presentation of rhabdoid tumors of the kidney. J Pediatr Hematol Oncol 23 (2): 105-8, 2001.
- Weeks DA, Beckwith JB, Mierau GW, et al. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms' Tumor Study Pathology Center. Am J Surg Pathol 13 (6): 439-58, 1989.
- Rorke LB, Packer R, Biegel J Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol 24 (1): 21-8, 1995.
- Biegel JA, Zhou JY, Rorke LB, et al. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59 (1): 74-9, 1999.
- Versteege I, Sévenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394 (6689): 203-6, 1998.
- Sévenet N, Sheridan E, Amram D, et al. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65 (5): 1342-8, 1999.
- Savla J, Chen TT, Schneider NR, et al. Mutations of the hSNF5/INI1 gene in renal rhabdoid tumors with second primary brain tumors. J Natl Cancer Inst 92 (8): 648-50, 2000.
- Parham DM, Roloson GJ, Feely M, et al. Primary malignant neuroepithelial tumors of the kidney: a clinicopathologic analysis of 146 adult and pediatric cases from the National Wilms' Tumor Study Group Pathology Center. Am J Surg Pathol 25 (2): 133-46, 2001.
- Jimenez RE, Folpe AL, Lapham RL, et al. Primary Ewing's sarcoma/primitive neuroectodermal tumor of the kidney: a clinicopathologic and immunohistochemical analysis of 11 cases. Am J Surg Pathol 26 (3): 320-7, 2002.
- Blakely ML, Shamberger RC, Norkool P, et al. Outcome of children with cystic partially differentiated nephroblastoma treated with or without chemotherapy. J Pediatr Surg 38 (6): 897-900, 2003.
- Wilms' tumor status report, 1990. By the National Wilms' Tumor Study Committee. J Clin Oncol 9 (5): 877-87, 1991.
- D'Angio GJ, Breslow N, Beckwith JB, et al. Treatment of Wilms' tumor. Results of the Third National Wilms' Tumor Study. Cancer 64 (2): 349-60, 1989.
- Ritchey ML, Coppes MJ The management of synchronous bilateral Wilms tumor. Hematol Oncol Clin North Am 9 (6): 1303-15, 1995.
- Ritchey ML, Pringle KC, Breslow NE, et al. Management and outcome of inoperable Wilms tumor. A report of National Wilms Tumor Study-3. Ann Surg 220 (5): 683-90, 1994.
- Green DM, Children's Oncology Group Phase III Multimodality Therapy Based on Histology, Stage, Age, and Tumor Size in Children With Wilms' Tumor, Clear Cell Sarcoma of the Kidney, or Rhabdoid Tumors of the Kidney, COG-Q9401, Clinical trial, Closed.
- Green DM, Breslow NE, Beckwith JB, et al. Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (1): 237-45, 1998.
- Green DM, Breslow NE, Beckwith JB, et al. Treatment with nephrectomy only for small, stage I/favorable histology Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 19 (17): 3719-24, 2001.
- Green DM, Breslow NE, Beckwith JB, et al. Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (1): 237-45, 1998.
- Green DM, Breslow NE, Beckwith JB, et al. Effect of duration of treatment on treatment outcome and cost of treatment for Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (12): 3744-51, 1998.
- Green DM, Children's Oncology Group Phase III Multimodality Therapy Based on Histology, Stage, Age, and Tumor Size in Children With Wilms' Tumor, Clear Cell Sarcoma of the Kidney, or Rhabdoid Tumors of the Kidney, COG-Q9401, Clinical trial, Closed.
- Green DM, Breslow NE, Beckwith JB, et al. Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (1): 237-45, 1998.
- Green DM, Beckwith JB, Breslow NE, et al. Treatment of children with stages II to IV anaplastic Wilms' Tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 12(10): 2126-2131, 1994.
- Green DM, Breslow NE, Beckwith JB, et al. Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (1): 237-45, 1998.
- Green DM, Breslow NE, Beckwith JB, et al. Effect of duration of treatment on treatment outcome and cost of treatment for Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (12): 3744-51, 1998.
- Green DM, Beckwith JB, Breslow NE, et al. Treatment of children with stages II to IV anaplastic Wilms' Tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 12(10): 2126-2131, 1994.
- Montgomery BT, Kelalis PP, Blute ML, et al. Extended followup of bilateral Wilms tumor: results of the National Wilms Tumor Study. J Urol 146 (2 ( Pt 2)): 514-8, 1991.
- Breslow NE, Takashima JR, Ritchey ML, et al. Renal failure in the Denys-Drash and Wilms' tumor-aniridia syndromes. Cancer Res 60 (15): 4030-2, 2000.
- Fuchs J, Wünsch L, Flemming P, et al. Nephron-sparing surgery in synchronous bilateral Wilms' tumors. J Pediatr Surg 34 (10): 1505-9, 1999.
- Ritchey ML, Green DM, Thomas PR, et al. Renal failure in Wilms' tumor patients: a report from the National Wilms' Tumor Study Group. Med Pediatr Oncol 26 (2): 75-80, 1996.
- Horwitz JR, Ritchey ML, Moksness J, et al. Renal salvage procedures in patients with synchronous bilateral Wilms' tumors: a report from the National Wilms' Tumor Study Group. J Pediatr Surg 31 (8): 1020-5, 1996.
- Wilms' tumor status report, 1990. By the National Wilms' Tumor Study Committee. J Clin Oncol 9 (5): 877-87, 1991.
- Ritchey ML The role of preoperative chemotherapy for Wilms' tumor: the NWTSG perspective. National Wilms' Tumor Study Group. Semin Urol Oncol 17 (1): 21-7, 1999.
- Zuppan CW, Beckwith JB, Weeks DA, et al. The effect of preoperative therapy on the histologic features of Wilms' tumor. An analysis of cases from the Third National Wilms' Tumor Study. Cancer 68 (2): 385-94, 1991.
:: Patología Neoplásica Urológica ::
Página: 43 de 58
- 43 -
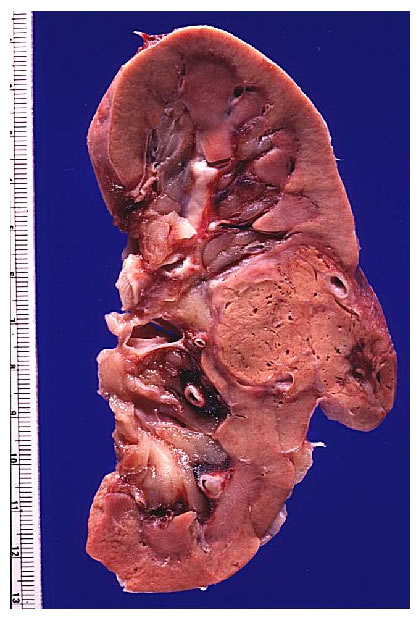
Fotografía: Ejemplo de Adenocarcinoma Renal
(Se sitúa en la porción media del riñón, es amarillo y presenta una zona central de necrosis y hemorragia)
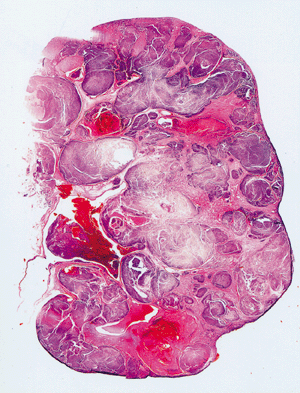
Fotografía: Carcinoma Renal.
(Renal carcinoma: the patient was operated upon by Wade on September 11th 1926. There was a recent history of painless haematuria. The histological diagnosis was recorded as `papillary carcinoma'. HE; approximately one half natural size.)
![]()
Fotografía: Adenocarcinoma Renal
(Es un adenocarcinoma renal de unos 8 cm., que afecta al polo inferior del riñón, y que presenta múltiples lóbulos, encapsulado, con extensiones que sobrepasan la capa cortical, pero que no afecta a la grasa perirrenal)
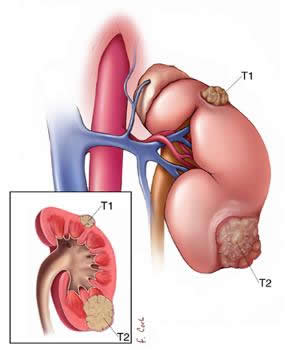
Esquema: pTNM - T1/T2
(Se observan las diferencias en cuanto a tamaño en dos tumores renales que afectan al mismo riñón, equivaliendo una a un T1 y la otra a un T2 según la clasificación pTNM)
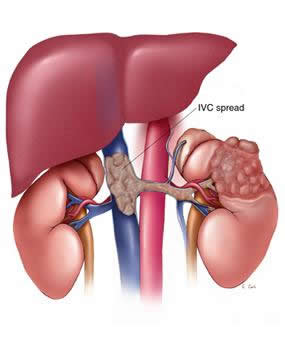
Esquema: pTNM - T3b
(En este caso la expansión del adenocarcinoma ha afectado a la vena renal y a la vena cava, lo cual nos permite englobarlo como T3b en la clasificación pTNM)
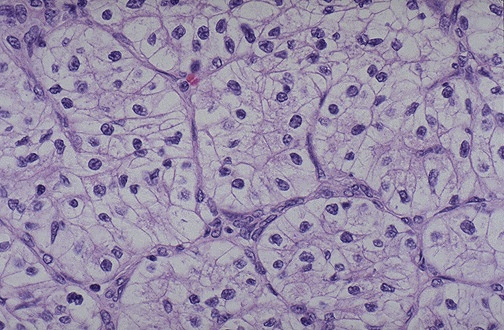
Fotografía: Adenocarcinoma Renal de Células Claras.
(Corte histológico H/E, en el que se observa el patrón característico de un adenocarcinoma renal de células claras: células grandes, citoplasma claro (Cargado de lípidos y glucógeno), y límites perfectamente definidos.)
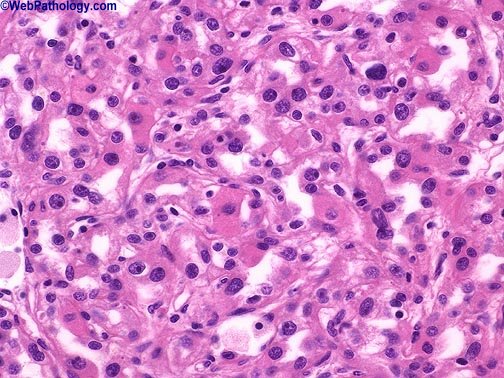
Fotografía: Carcinoma de Células Renales - G3
(Corte histológico H/E, donde vemos un grupo de células compatible con un Grado 3 de Fuhrman, los núcleos son más irregulares, con un diámetro de unas 20 µm, y el nucleolo se ve perfectamente en muchas de ellas .)
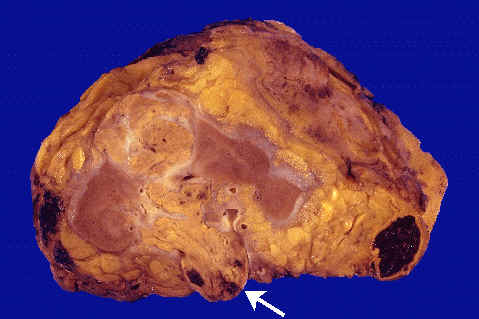
Fotografía: Carcinoma de Células Renales
(Imagen macroscópica de un adenocarcinoma renal o carcinoma de células renales que por su tamaño podría ser compatible con un Estadio III de la AJCC, ya que presenta una clara invasión de la vena renal., lo que representa un T3 en la clasificación pTNM.)
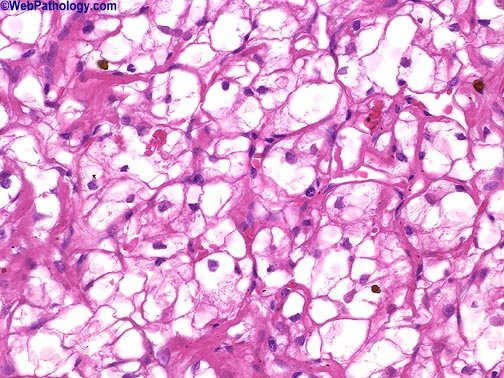
Fotografía: Carcinoma de Células Renales - G1
(Corte histológico H/E, donde vemos un grupo de células compatible con un Grado 1 de Fuhrman, con un núcleo redondo y uniforme.)
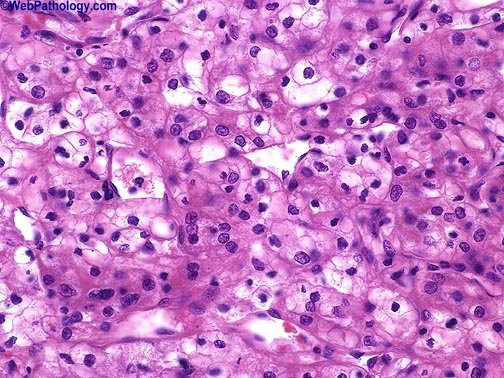
Fotografía: Carcinoma de Células Renales - G2
(Corte histológico H/E, donde vemos un grupo de células compatible con un Grado 2 de Fuhrman, con un núcleos más irregulares que en G1, con un diámetro de unas 15 µm, y nucleolo visible a grandes aumentos.)

Fotografía: Adenocarcinoma Renal - Cambio Sarcomatoide.
(Corte histológico H/E (en blanco y negro), donde se muestra una celularidad de aspecto fusiforme con haces entrelazados típico de un cambio sarcomatoide en un carcinoma de células renales.)

Fotografía: Adenocarcinoma Renal de Células Claras.
(Aspecto macroscópico de un adecnocarcinoma renal de células claras, en el que son evidentes los rasgos más caracteríticos de esta variante de adenocarcionma que supone más del 80% de los tumores malignos de riñón, como son: localización periférica, bien delimitado, seudoencapsulado, zonas necróticas, hemorrágicas, etc..)

Fotografía: Adenocarcinoma Renal - Resección.
(La masa renal ha sido reseccionada y la fotografía muestra todas las características de este tipo de adenocarcinomas, con zonas amarillas, necrosis, zonas quísticas, etc. Además se deberá hacer un estudio de los márgenes de resección para comprobar si están afectados o no a nivel microscópico.)

Esquema: Anatomía Renal Normal
(Se observa la ubicación anatómica de los riñones en el abdomen, y sus relaciones con la arteria y vena renales, ureter, arteria aorta abdominal, vena cava inferior. Además se observa su estructura básica: Corteza, Médula, Cálices, Pelvis, etc...)

Fotografía: Ejemplo de Adenoma Cortical.
This 29 year old female presented with virilizing symptoms and was found to have a large adenoma in the right adrenal. The tumor is composed of compact cells arranged in nests and cords.

Fotografía: Ejemplo de Adenoma Cortical.
Adrenal cortical adenoma removed from a 35 year old male who presented with Cushing's Syndrome. The tumor is bright yellow and grossly resembles zona fasciculata seen in the specimen.

Fotografía: Fibroma Renal.
Usually found incidentally as a small well-circumscribed whitish nodule in the renal medulla

Fotografía: Fibroma Renal.
Microscopically, it consists of stellate cells in loose myxoid stroma. A few entrapped tubules may be present.

Fotografía: Angiolipoma.
This gross photograph shows angiomyolipoma arising in the superior pole of right kidney. About 50% of the cases are associated with tuberous sclerosis. The rest occur sporadically. Tuberous sclerosis-associated tumors tend to be multiple, bilateral, and small & are usually asymptomatic

Fotografía: Angiolipoma.
The smooth muscle bundles are often seen arising from the vessel wall. The blood vessels are thick-walled with small abnormal or abortive lumens. Cytologic atypia may be prominent. The smooth muscle cells react with HMB-45

Fotografía: Angiolipoma.
Microscopically, angiomyolipomas are composed of variable amounts of adipose tissue, smooth muscle, and thick-walled blood vessels

This low-power micrograph from a section near the central scar shows nests of tumor cells scattered in abundant loose myxoid stroma

The classic histologic pattern of oncocytoma consists of nested or organoid arrangement of tumor cells surrounded by moderate to abundant quantity of myxoid or hyalinized stroma

A number of atypical features have been described in the histologic spectrum of renal oncocytomas. Infiltration of perinephric fat was seen in 11% of cases in one study (Amin MB et al. Am J Surg Pathol 1997 Jan; 21(1):1-12.)

Carcinoma de Células Renales Cromófobo
Grossly, chromophobe renal cell carcinoma is usually a well-circumscribed solitary mass in the kidney as seen in this case. The cut surface may be yellow, beige, or brown with close resemblance to oncocytoma. A central scar is seen in this specimen. Case contributed by: Liang Cheng, MD, Dept. of Pathology, Indiana University School of Medicine, Indianapolis.

The nuclei are moderately sized and hyperchromatic. Bi- and multinucleated cells are not uncommon. Chromophobe RCC appears to have a better 5-yr disease-specific and progression-free survival than conventional (clear cell) RCC or papillary RCC. Am J Surg Pathol 2002 March; 26(3) 281-91

The cytoplasmic density is greater at the periphery of the tumor cells in chromophobe RCC making the cytoplasmic borders thick and distinct

Esquema: Tumor de Wilms.
(Representa un Tumor de Wilms o Nefoblastoma localizado en el riñón derecho de un niño, siendo la tumoración renal que con mayor frecuencia aparece en la infancia.)

Fotografía: Tumor de Wilms.
(El Tumor de Wilms es la 5 neoplasia más frecuente en pediatría y debido a que su origen es el tejido renal embrionario podemos encontrar elementos epiteliales, del estroma o blastemáticos)
Descargar
| Enviado por: | LordFaltian |
| Idioma: | castellano |
| País: | España |
Todos los derechos reservados.