Biología
Bioquímica
Introducción
TEMA 1
INTRODUCCIÓN A LA BIOQUÍMICA
La bioquímica tiene como objetivo más importante el estudio de la estructura, organización y funciones de la materia viva desde el punto de vista molecular.
Según los aspectos tratados, esta puede dividirse en 3 bloques:
1.- bioquímica estructural
2.- bioquímica metabólica o metabolismo
3.- biología molecular
La bioquímica estructural estudia la composición, conformación, configuración y estructura de las moléculas de la materia viva, relacionándolas con su función bioquímica.
El metabolismo estudia las transformaciones, funciones y reacciones químicas que sufren o llevan a cabo las moléculas de la materia viva.
La biología molecular estudia la química de los procesos y moléculas implicadas en la transmisión y el almacenamiento de la información biológica.
La bioquímica, fundamentalmente extrae conocimientos de biología, química y genética, y usa técnicas que ha importado de la física.
Origen de la bioquímica
El origen del término “bioquímica” se remonta a los comienzos del siglo XIX, cuando imperaba una teoría conocida como “vitalismo” que sostenía que las sustancias existentes en la materia viva eran cualitativamente diferentes de la materia no viva, y que no se comportaban según las leyes conocidas de la física y la química.
Esto se derrumbó a raíz de los trabajos de un bioquímico alemán (Whöler en 1828), que sintetizó en un laboratorio urea a partir de cianato amónico. Hasta entonces se pensaba que solo se podía sintetizar en los animales, en el riñón.
La corriente vitalista aún persistió porque se creía que las reacciones de la materia viva solo podían realizarse en células vivas. Según el vitalismo, las reacciones biológicas se producían por una fuerza vital de naturaleza misteriosa pero no por procesos químicos o físicos.
Se encargaron de echar esto abajo los hermanos “Buchner” en 1897, observando que extractos de células de levaduras rotas y por lo tanto muertas, eran capaces de llevar a cabo la fermentación del azúcar hasta etanol. Esto abrió el camino al estudio de las reacciones y procesos bioquímicos “in vitro”. A partir de aquí se avanzó más rápidamente en el conocimiento de las diferentes rutas metabólicas.
A finales del siglo XIX todavía persistían científicos vitalistas sosteniendo que la naturaleza de la catálisis biológica (las enzimas) y sus estructuras eran demasiado complejas para describirlas en términos químicos.
Este último postulado se derrumbó en 1926, cuando Sumner fue capaz de cristalizar una proteína (ureasa purificada de la judía) demostrando que aunque las proteínas tienen unas estructuras grandes y complejas, es posible sintetizarlas como otro compuesto inorgánico cualquiera y que sus estructuras pueden determinarse con los métodos de la física y química.
A partir de aquí, la contribución más importante consistió en establecer las estructuras químicas básicas de las sustancias biológicas, identificar las reacciones de cada ruta metabólica y localizar éstas en el interior de la célula.
Esto es apoyado por el avance en las técnicas de la biología celular.
En la mitad del siglo XX se desarrolla la biología molecular, aunque la idea de gen ya había sido propuesta en el siglo XIX por Mendel.
A mediados del siglo XX todavía nadie había aislado un gen ni se había determinado su composición química. La mayoría de los científicos pensaban que los ácidos nucleicos tenían una función meramente estructural, y que tan solo las proteínas eran lo suficientemente complejas estructuralmente para ser portadoras de la información genética.
En los años 40 y 50 se demostró que esto era erróneo, y que el ADN era el portador de la información genética.
Watson y Crick publicaron la estructura en doble hélice del ADN, así como todos lo descubrimientos posteriores relacionados con los mecanismos de la transmisión de la información biológica.
Esto dio lugar a una nueva rama de la biología, la biología molecular.
La bioquímica también nutre de conocimientos a otras ramas de la biología como la biofísica, la nutrición, investigación médica o biotecnología entre otros.
PROPIEDADES, ORIGEN Y EVOLUCIÓN DE LAS BIOMOLÉCULAS
Bioelementos o elementos biogenéticos:
Casi todos los bioelementos se sitúan en la primera mitad del sistema periódico. La composición en elementos de casi todos los seres vivos es básicamente similar, aunque constan de diferencias como el caso del yodo, que es imprescindible para los vertebrados pero no para otros animales.
En el hombre se han identificado al menos 30 que son indispensables en proporciones muy diversas. Según su abundancia, se clasifican en 3 grandes grupos:
-
Bioelementos primarios
-
Secundarios
-
Oligoelementos
Los bioelementos primarios son H, O, C, N. Son los más abundantes en todo el organismo. En el caso del hombre, el 99% del total.
Los bioelementos secundarios son Ca, P, K, S, Na, Cl, Mg, Fe. Son mucho menos abundantes, pero también están presentes en todos los organismos. En el cuerpo humano constituyen el 0,7% del total de átomos.
Los oligoelementos son el grupo más numeroso: Mn, I, Co, Cu, Zn, F, Mo, Se....
Aparecen tan solo en cantidades trazas, y la diferencia con los anteriores es que algunos no son esenciales en todos los organismos.
La ausencia de estos bioelementos determina la aparición de enfermedades.
La razón por la cual los primarios están en mayor proporción se debe a que son los elementos más pequeños del sistema periódico que pueden formar enlaces covalentes.
En el caso concreto del C, este presenta la característica de que además de formar enlaces de este tipo con otros átomos de otros elementos, también puede formar enlaces covalentes consigo mismo, dando lugar a cadenas de átomos de carbono que pueden tener más de 100 átomos de C y que pueden ser lineales, ramificadas o cíclicas.
Los enlaces pueden ser dobles, lo que da lugar a la aparición de una gran variedad de grupos funcionales.
También los enlaces pueden ser triples, aunque son muy raros.
El silicio es un átomo que también puede formar cadenas consigo mismo, pero estos enlaces se oxidan y se rompen por lo que en una atmósfera de como la nuestra, éstos son inestables.
Las funciones desempeñadas por los bioelementos son muy variadas, y las agruparemos en 3 tipos fundamentales: plásticas / estructurales, catalíticas y osmóticas.
Hay elementos que desempeñan funciones plásticas o estructurales ya que forman parte de huesos, tejidos fibrosos, tegumentos, etc... Dentro de este grupo están C, O, H, N, pero también S, P, Ca.
En segundo lugar, muchos desempeñan funciones catalíticas. Un ejemplo es el caso del Fe, que participa en el transporte de oxígeno y electrones, o el caso del Zn que es cofactor de muchas encimas, o el I, que forma parte de las hormonas tiroideas o el Co, que forma parte de la vitamina .
En tercer lugar hay bioelementos con funciones osmóticas. Destacan el Na, K, Cl, que en forma iónica intervienen en procesos como la distribución del agua en compartimentos intra y extracelulares en procesos como el mantenimiento de potenciales de membrana, así como otros relacionados.
Del mismo modo que los bioelementos, definimos las biomoléculas o principios inmediatos como aquellas constituyentes de los seres vivos, que según su naturaleza las clasificamos en 2 grupos, inorgánicas y orgánicas.
Dentro de las inorgánicas, tenemos el , gases como y , sales inorgánicas como aniones (como fosfato, bicarbonato) o como cationes (como el amonio).
Dentro de las moléculas orgánicas tenemos además de glúcidos, lípidos, proteínas y ácidos nucleicos, otras amplias variedades de biomoléculas que se conocen en conjunto como “metabolitos”, intermediarios de etapas de rutas metabólicas, como el piruvato, ácido cítrico, urea, etc.
Aunque existe una gran variedad según los distintos tipos de células, el principal componente celular siempre es el , que representa el 75% aproximadamente. El 2º componente celular en abundancia son las proteínas, con un 15%. El 10% restante se reparte entre las demás biomoléculas. Aproximadamente, un 2% de azúcares, 3% de lípidos, un 2% de ARN, un 0,5% de ADN, un 1,5% de metabolitos intermediarios y un 1 % de sales. Hay ciertos grupos de células que se salen de esta generalidad debido a sus funciones, como las células adiposas, que contienen un % mayor en lípidos, o las células hepáticas, con un % mayor en azúcares. Estos porcentajes también pueden variar según el estado fisiológico de la célula. Por ejemplo, si la célula se encuentra en mitosis, el % será mayor en ácidos nucleicos, que si estuviese madura.
Tipos de enlaces entre biomoléculas
Intramoleculares: unen los átomos para formar la biomolécula. Pueden ser covalentes o iónicos.
Intermoleculares : las moléculas interaccionan formando asociaciones supramoleculares que pueden llegar a ser bastante complejas. Las fuerzas intermoleculares son más débiles que las covalentes. Entre estas, tenemos las electrostáticas, puentes de hidrógeno, fuerzas de Van der Waals, interacciones hidrofóbicas y polares.
Ejemplos típicos de estas estructuras supramoleculares son los ribosomas, la cromatina, las membranas...
Modelos moleculares
Son modelos creados que usamos para representar la estructura en 3D de las biomoléculas. Esta la podemos representar por 3 tipos de modelos: espaciales, de esferas y varillas, y esqueléticos.
Los primeros son los más realistas, ya que en ellos el tamaño y configuración de un átomo en concreto viene determinado por las propiedades del enlace y por los radios de Van der Waals.
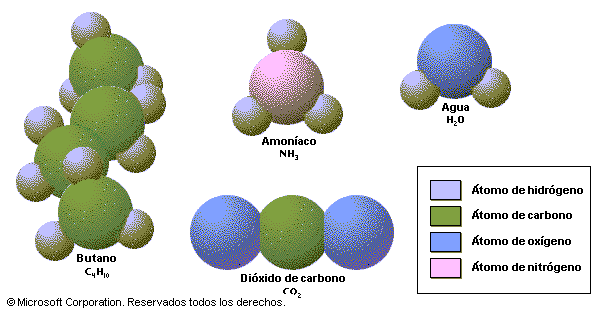
Ejemplos de espacial
Los modelos de esferas y varillas son menos realistas ya que los átomos están representados por esferas más pequeñas de lo que les correspondería según los radios de Van der Waals, pero en ellas se aprecia más fácilmente la disposición de los enlaces, representados por las varillas.
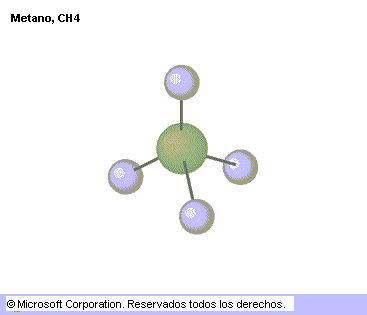
Ejemplos del modelo de representación con esferas y varillas
Los esqueléticos nos proporcionan la imagen más simple ya que solo muestran la disposición molecular. Los átomos no están indicados explícitamente sino que la disposición de estos se deducen en los sitios de unión de los enlaces y sus términos. Estos modelos se usan sobre todo para representar grandes moléculas que pueden tener varios miles de átomos.
Los átomos de H solo se suelen representar en el primer tipo de modelos.
Además de estos 3 modelos también se suelen utilizar representaciones esquemáticas de estructuras extensas como sucede con la clásica representación del ADN como 2 cintas en
espiral o como sucede también en el caso de las proteínas en sus modelos de cintas. Las cintas representarían las regiones de la proteína en hélice , las flechas en lámina , y los cordones como estructuras enrolladas al azar.
TEMA 2
EL AGUA EN LOS PROCESOS BIOLÓGICOS
Estudiaremos el porque es la molécula más abundante y porque la inmensa mayoría de las reacciones bioquímicas tienen lugar en un medio acuoso y obedecen a las leyes físico-químicas de las disoluciones acuosas.
-
En primer lugar, el es líquida en un amplio rango de temperaturas (0-100ºC). Esto permite que la vida pueda desarrollarse en condiciones climáticas muy dispares.
-
Presenta una densidad máxima a 4ºC. En consecuencia, el hielo flota sobre el líquida, actuando como aislante térmico, lo que permite que la gran masa de los océanos se mantenga líquida y por tanto, la vida de numerosos seres acuáticos.
-
Elevada constante dieléctrica, que permite la disolución de la mayoría de las sales minerales, ya que debilita las fuerzas electrostáticas entre los iones.
-
Es un dipolo, por lo tanto, sus moléculas tienden a rodear a los iones aislándolos unos de otros, facilitando la disolución de las sales. se dice que estos iones quedan solbatados. El también solbata otras moléculas polares no iónicas como el etanol.
-
Elevado calor específico, gracias a lo que los animales pueden ganar o perder bastante calor con escasa modificación de la temperatura corporal. Esto explica el papel termorregulador corporal de la sangre.
-
Elevado calor de vaporización. Esto quiere decir que un animal puede eliminar el exceso de calor vaporizando cantidades de relativamente pequeñas a través de pulmones y piel, pudiendo mantener así la temperatura corporal por debajo de la ambiente.
-
Elevada tensión superficial. Sus moléculas se ordenan de modo que la superficie libre sea mínima. Las sustancias denominadas tensoactivas, como los detergentes, son capaces de disminuir la tensión superficial del agua, lo que facilita la mezcla o inclusión de las grasas en un medio acuoso. Así es como actúan las sales del intestino delgado, que facilitan la disolución de las grasas en este.
-
Una disociación en y crítica para muchos procesos biológicos. Las células vivas tienen mecanismos bastante eficaces para controlar estas concentraciones.
Muchas de estas propiedades se deben a que sus moléculas se hayan enlazadas por fuerzas intermoleculares que son especialmente, puentes de H y fuerzas de Van der Waals.
Disoluciones acuosas
Una disolución es una mezcla homogénea de moléculas diferentes (solutos) en un disolvente. En bioquímica la gran mayoría de las disoluciones son acuosas. Al disolverse en agua, estos solutos modifican las propiedades del disolvente como el color, densidad...
Algunas de estas propiedades no dependen de la naturaleza del soluto, sino solo del número de partículas disueltas y las denominamos propiedades coligativas de las disoluciones, que son:
-
descenso de la P de vapor
-
descenso crioscópico, o de la temperatura de congelación
-
incremento de la temperatura de ebullición
-
P osmótica
Desde el punto de vista fisiológico destaca la presión osmótica, porque las membranas celulares actúan como membranas semipermeables.
Ejemplo: los eritrocitos, que forman parte de la sangre. Si colocamos uno en agua destilada, este estallará (se lisará) ya que en su interior hay una concentración de soluto, y el agua entrará en su interior hasta equilibrar las concentraciones interna y externa. Se calcula que la presión a soportar por las paredes del eritrocito sería de 7,6 atmósferas.
Cuando forman parte de la sangre no se lisan porque este medio es isotónico respecto del interior del glóbulo rojo.
Para mantener constante la concentración de solutos en el medio interno del organismo, hay diversos mecanismos. Destacan la sed y la filtración renal, reguladas por osmoreceptores conectados con el sistema nervioso central.
Las disoluciones acuosas las diferenciamos en dos tipos: moleculares e iónicas.
En las primeras, las moléculas disueltas mantienen su integridad como en el caso de azúcares, proteínas... En las iónicas, las moléculas disueltas se disocian en el disolvente en dos o más iones.
Que un disolución sea una o otra repercute directamente en sus propiedades coligativas, ya que dependen del número de partículas disueltas, y por tanto, como en una disolución iónica, el número de iones es mayor que el de moléculas disueltas, el efecto sobre las propiedades coligativas será mayor, mientras que en el caso de las disoluciones moleculares será mucho menor a causa de que las partículas son tantas como las moléculas disueltas.
Un tercer tipo de disoluciones son las coloidales, que se caracterizan porque las partículas del soluto superan un determinado tamaño, (normalmente un diámetro mayor a 10 o una masa superior a 10 kilodalton). Las disoluciones coloidales difieren de las disoluciones verdaderas en algunas propiedades. Por ejemplo, se pueden precipitar por centrifugación a alta velocidad. La superficie de contacto de las moléculas con el agua o interfase adquiere una especial relevancia en este caso, y numerosas técnicas analíticas se basan en las propiedades de estas interfases.
Biomoléculas que dan lugar a coloides son los polisacáridos, proteínas y ácidos nucleicos. Estas macromoléculas presentan ,en su interfase con el agua, grupos polares como el OH o iónicos como el carboxilo o el amino, que ordenan a su alrededor una gran cantidad de moléculas de agua. Como estas macromoléculas son de gran tamaño, incluso las disoluciones concentradas contienen un número pequeño de moléculas, por lo tanto las propiedades coligativas de las disoluciones coloidales son poco observables. Sin embargo, la P osmótica sí es importante, y presenta algunas peculiaridades en los siguientes casos:
-
cuando el coloide es iónico
-
cuando el compartimento que contiene el coloide está limitado por una membrana semipermeable
-
cuando al otro lado de la membrana semipermeable existe un soluto iónico difusible
En estos casos el coloide sí origina una presión osmótica que se debe, por una parte, a la presencia de las macromoléculas coloidales, pero también a la existencia en este compartimento de un exceso de partículas iónicas difusibles que son atraídas por el coloide. A esta presión se le denomina presión coloidosmótica, debida a que el coloide tiene carga y por lo tanto atrae a iones que son los que crean la P.
Esto sucede en biología en muchos lugares como entre el plasma y el líquido intersticial, separados por el endotelio del capilar.
Las moléculas que se disocian decimos que son electrolitos y diferenciamos
-
los electrolitos fuertes, cuando la disociación es total,y
-
los débiles, cuando la disociación es solo parcial
Equilibrios iónicos
El agua es un electrolito débil que se disocia en muy baja proporción de la siguiente manera: . Solamente una molécula de cada está disociada, y esto presenta un gran interés en biología.
. En agua pura, , es decir, que el agua pura tiene un PH = 7.
Cualquier sustancia que haga variar una de las concentraciones de los iones hará que la concentración del otro tipo iónico se modifique de modo que el producto siga siendo constante.
El interés fisiológico del PH se refleja en varios aspectos. En primer lugar, el funcionamiento armónico de muchos procesos del organismo requiere la actuación concertada de muchas encimas, y la acción catalítica de estas dependen del PH. Además en el interior de las células se producen gradientes locales de PH que se usan en el almacenamiento de energía química en forma de ATP, como sucede por ejemplo entre la mitocondria y el citosol.
Por otra parte, proceso como el intercambio de gases en los pulmones está regulado por cambios muy suaves de PH en el interior del eritrocito.
En resumen, en bioquímica tendremos que tener en cuenta el tipo de disolución que usemos y el PH.
La mayoría de los ácidos y bases que tienen interés fisiológicos son, al igual que el agua, electrolitos débiles y están solo parcialmente disociados. Por ejemplo, el ácido acético se va a disociar según esta ecuación: . En el equilibrio, la constante de equilibrio es: . Igualmente sucede con otros ácidos y bases débiles.
Para los ácidos y bases débiles, K es muy baja y con lo que se trabaja es con el pK (-log K). En la escala de pK los ácidos débiles tienen valores menores a 7 y las bases mayores a 7. Cuando el ácido se encuentra disociado al 50% la concentración de la forma no disociada es igual a la concentración de la forma disociada y por lo tanto podemos decir que PH=pK . También podemos definir el pk como el PH al que el 50% del ácido está disociado.
El pK de los ácidos y bases débiles se puede determinar mediante las curvas de titulación. Lo más importante de las curvas es que muestran como un ácido débil y su anión pueden actuar como tampones.
Un sistema tampón o amortiguador es usado por el organismo para protegerse de variaciones bruscas de PH.
NOTA: además de tampones o amortiguadores podemos encontrarnos con el término inglés “Buffers”.
Estas disoluciones contienen en proporciones análogas las formas disociada y no disociada de un ácido o base débil, que conseguimos mezclando en proporciones adecuadas un ácido débil y base fuerte, o base débil y ácido fuerte, de forma que lo que se origina es una mezcla de un electrolito y una sal fuerte. Cualquier factor externo que tienda a variar la concentración de desplazará el equilibrio hasta que la nueva concentración de sea similar a la inicial. A efectos cualitativos, los sistemas tampón se rigen por la ecuación de Héndersson - Hasselbach, que es .
Las disoluciones reguladoras cumplen una serie de normas:
El PH de una reguladora no depende de las concentraciones absolutas de ácido y base o sal si no de la proporción entre las formas disociada y no disociada, con lo que a una disolución tampón podemos añadir agua sin modificar su PH. Aunque no varía, lo que ocurre es que las disoluciones más concentradas tienen mayor capacidad de tamponamiento. Además la amortiguación es máxima cuando el PH del medio coincide con el pK del amortiguador. Si la diferencia entre el PH y el pK es mayor a 2 entonces no hay capacidad amortiguadora.
Una base débil actuará como tampón al añadir un ácido fuerte siempre que al reaccionar entre sí quede algún exceso de base.
En el organismo tenemos una serie de amortiguadores fisiológicos, de los cuales destacan los fosfatos. En los tejidos el fosfato se haya combinado con azúcares, lípidos o ácidos nucleicos. El ácido fosfórico tiene 3 átomos de H disociables. Su disociación es gradual y cada uno de ellas posee un pK específico. Pues bien, solo la disociación del segundo protón tiene interés fisiológico ya que su pK es 6,86 , cercano al PH fisiológico.
Otro importante es el sistema del bicarbonato, ya que el ácido carbónico se haya en equilibrio con el y por tanto la concentración de ácido carbónico en sangre se modifica variando la ventilación pulmonar. Por último, el amortiguador más importante fisiológicamente son las proteínas, que contienen grupos funcionales que son ácidos y bases débiles con una amplia gama de pKs, de manera que funcionan como buenos amortiguadores casi a cualquier PH. Por su abundancia en la sangre y su papel en la respiración, la hemoglobina destaca como amortiguador entre las proteínas. Su peculiaridad más importante en este sentido es que su pK varía según esté o no oxigenada, lo que le confiere una capacidad amortiguadora adicional.
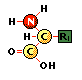
los aminoácidos, constituyentes de proteínas poseen un grupo carboxilo y otro amino.
Metodología bioquímica
TEMA 3
EL MATERIAL BIOLÓGICO. HOMOGENADO DE TEJIDOS
Aspectos generales de la metodología bioquímica
En la investigación bioquímica con frecuencia se necesita purificar un componente particular de entre una mezcla compleja. Diferenciamos 2 separaciones, las analíticas y las separativas.
En las analíticas el objetivo es identificar y estimar pequeñas cantidades de los compuestos, y generalmente no es necesario recuperar el compuesto después del proceso de separación . Para determinar la presencia de un compuesto en una mezcla se compara su comportamiento durante el proceso analítico con el de un compuesto conocido que se denomina “estándar” o “patrón”, y que se somete exactamente al mismo tratamiento.
La determinación cuantitativa del compuesto puede hacerse sin necesidad de purificación previa en algunos casos, en los que dicho componente posee una proporción única que lo caracteriza, como sucede con los ensayos de actividad encimática en extractos crudos o células enteras.
En cuanto a las separativas, el objeto es distinto. Se trata de aislar y recuperar una gran cantidad del compuesto en un grado de pureza para poder, con este compuesto aislado, estudiar sus propiedades químicas o biológicas. Las separativas requieren una mayor cantidad de material de partida que las analítica para garantizar un alto grado de recuperación del compuesto. Los métodos utilizados poseen una resolución menor que los métodos usados en las técnicas analíticas.
Usualmente los procesos de purificación de una molécula en bioquímica comienzan con técnicas preparativas que nos dan una buena cantidad de material ya purificado. A continuación se sigue purificando hasta que alcancemos el grado de pureza deseado. Normalmente a mayor pureza menor recuperación.
El material biológico usado en bioquímica
La experimentación en bioquímica puede llevarse a cabo de dos modos fundamentales, “in vivo” e “in vitro”.
El primer término se aplica a organismos multicelulares y utiliza plantas o animales enteros o también parte de los animales en las cuales determinados órganos están prefundidos con nutrientes. La perfusión implica el bombeo de una disolución isotónica cargada con nutrientes, fármacos u hormonas a través del órgano. Esta solución asume el papel de la circulación normal, aportando nutrientes y eliminando sustancias de desecho.
Los animales se utilizan en investigación fundamental así como en médica, veterinaria y en pruebas toxicológicas. Los animales más empleados son ratones, ratas y cobayas, debido a su bajo precio y facilidad de manejo, pero también otros animales como conejos, muy empleados para preparar anticuerpos. También son utilizados gatos, perros, monos, cerdos, etc...
Estos animales deben mantenerse en pequeños grupos homogéneos en lo relativo a edad, sexo, estado fisiológico... y controlados desde el punto de vista de nutrición, posibles enfermedades...
Los resultados extraídos con experimentos con animales deben ser sometidos a la estadística, realizando experimentos con más animales.
Las plantas enteras se han necesitado en investigación básica para el estudio de procesos como la fotosíntesis, respiración, asimilación del nitrógeno... pero también para investigación aplicada en campos como loa patología, polución, tecnología de los alimentos...
Las plantas, según las necesidades del experimento y especie, pueden obtenerse de plantaciones de exterior o de invernadero. La utilización de estos últimos tiene la ventaja de que se controlan aspectos como la temperatura, humedad, iluminación...
Los “in vitro” implican la incubación del material biológico en ambientes físico - químicos artificiales. Este término se aplica a preparaciones encimáticas, órganos prefundidos pero aislados del animal, microorganismos intactos y a partes escindidas de animales o plantas.
También se usa en la experimentación in vitro células desagregadas de tejidos animales por tratamiento con encimas como la tripsina o colagenasa, que degradan la matriz extracelular que mantiene los tejidos y órganos unidos.
En un órgano normalmente coexisten varios tipos de células. Para separar estos tipos se usan dos métodos fundamentalmente. Por una parte la centrifugación que separa las células en función de su tamaño. Más recientemente se desarrollaron los denominados clasificadores de células activadas por fluorescencia, que consisten en que una suspensión celular se trata con una anticuerpo marcado con fluorescencia (unido a un componente fluorescente) que se dirige contra un antígeno de la superficie celular que está presente en cantidades diferentes en los distintos tipos de células. A continuación las células can pasando en fila de una a una por un rayo láser y se van separando físicamente de acuerdo con la cantidad de fluorescencia que presenta cada una. Estos aparatos son capaces de separar varios miles de células por segundo.
Además, para la experimentación in vitro también se pueden usar las células animales o vegetales en cultivo. La ventaja de la utilización de los cultivos es que obtenemos poblaciones homogéneas y además en condiciones controladas de manera que podemos inducir o reprimir un determinado encima o ruta metabólica.
En general, podemos decir que los métodos de esta investigación han permitido avanzar más rápido en los experimentos bioquímicos, aunque recibe una dura crítica ya que no siempre sucede lo mismo cuando estas células están en el laboratorio en cultivo que cuando están en vivo en el órgano.
Técnicas de homogenado de tejidos y fraccionamiento de orgánulos celulares
El primer paso en cualquier proceso analítico en el cual el contenido celular deba ser liberado, consiste en la homogeneización de tejidos y la rotura de celular. Esto nos proporciona una mezcla cruda que se puede utilizar como material de partida en procesos analíticos, o para la determinación de actividades encimáticas o estudios metabólicos cuando la incorporación de un determinado metabolito por el tejido intacto sea difícil. También podemos usar otros tejidos homogeneizados para la separación de los componentes celulares (fraccionamiento celular), en experimentos de compartimentación metabólica ( determinar en qué parte de la célula se produce un proceso o ruta metabólica).
El material biológico que se va a homogeneizar se debe seleccionar de modo que sea rico en los orgánulos de interés, y además muy activo en la vía metabólica que se va a estudiar.
Por ejemplo, si queremos estudiar la cadena respiratoria, seleccionaremos un tejido rico en mitocondrias, como el hígado fresco. Si quisiésemos aislar núcleos, tomaríamos un trozo de timo. Además tenemos que tomar la precaución especial de que la solución en la que se lleva a cabo la homogeneización debe ser isotónica (generalmente disoluciones de sacarosa). El fraccionamiento celular se hace mediante centrifugación diferencial ( a diferentes velocidades y durante tiempos distintos), así es posible separar núcleos, cloroplastos, lisosomas, mitocondrias, microsomas. El contenido del citosol permanece en el sobrenadante de la última centrifugación.
Muchas encimas y proteínas son termolábiles (se desnaturalizan por acción de la temperatura) por lo tanto estos procesos de homogeneización deben realizarse en frío (generalmente a 4 ºC en cámaras frías).
Los métodos empleados para la disyunción de las células se basan fundamentalmente en que en general, las células cultivadas son fáciles de romper. Esto se consigue por choque osmótico, ciclos de congelación - descongelación, o por digestión con encimas (proteasas y lipasas).
Otra manera de romper las células animales es el caso de disolventes orgánicos como el tolueno . sin embargo, levaduras y células vegetales poseen un pared celular resistente y dura que requiere que para romperla se combinen métodos encimáticos y mecánicos. Los métodos encimáticos implican el tratamiento de las células vegetales con pectinasas y celulasas. En el caso de levaduras su pared se rompe con la lisocima (mezcla de encimas).
Entre los métodos físicos tenemos:
-
molido con mortero ( en presencia de un agente abrasivo como alúmina)
-
agitación en presencia de pequeñas bolas de vidrio (es muy usado en levaduras, pero en las plantas se pueden dañar los orgánulos)
También se utilizan homogeneizadores que son sistemas de extrusión o prensas ( la french es la más usada con levaduras).
Otra manera es la ruptura celular por ultrasonidos. Es muy empleada con bacterias y su pared. Los ultrasonidos son eficaces pero presentan un inconveniente, la muestra se calienta mucho, con lo que debemos actuar de manera esporádica y enfriando la muestra entre aplicación y aplicación.
El material biológico lo conservamos con la técnica del frío, que es la más utilizada, congelando la muestra en nitrógeno líquido, porque se considera que las reacciones bioquímicas se detienen a -130 ºC, y el punto de ebullición del nitrógeno es de -196 ºC, con lo que nos asegura la baja temperatura requerida. Normalmente esta congelación se hace en presencia de un componente protector, que suele ser glicerol, que evita la formación intracelular de cristales de hielo como consecuencia de la congelación rápida que al descongelar provocaría la lisis de las membranas celulares.
Precipitación fraccionada y centrifugación. Ultracentrifugación.
La precipitación fraccionada es una fase preparativa de la purificación de proteínas. Se realiza con sales, comúnmente sulfato amónico.
Se basa en que en soluciones salinas concentradas, unas proteínas son más solubles que otras, y así podemos separar distintas fracciones proteicas.
El objetivo de la centrifugación es acelerar la sedimentación de sustancias químicas, células, partículas... creando campos gravitatorios artificiales mucho más intensos que el terrestre. Estos campos gravitatorios artificiales se consiguen haciendo girar la muestra a gran velocidad en unos aparatos llamados “centrífugas”. Las centrífugas de uso habitual alcanzan entre 3.000 G y 10.000 G , siendo 1G = aceleración de la gravedad terrestre.
En cuanto a las “ultracentrífugas”, estas alcanzan 400.000 G debido a su mayor velocidad de rotación y a su actuación en vacío.
En las centrifugaciones los tubos que contienen la muestra se colocan en una pieza que gira y que se llama “rotor”.
Para la centrifugación es esencial que el centro de gravedad del rotor coincida con el eje de rotación. Para ello, es imprescindible que los tubos con la muestra se coloquen equilibrados en el rotor, situándolos simétricamente con pesos iguales.
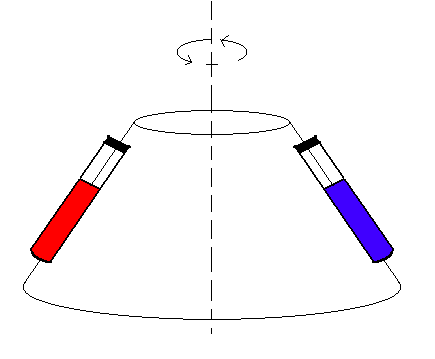
Rotor de ángulo fijo.
Mediante la centrifugación las partículas se separan en función de una característica que es el coeficiente de sedimentación, simbolizado por “S” y sus unidades son los “svedverg”, siendo 1 S = segundos. “S” aumenta con la masa de las partículas pero la relación no es lineal. Podemos usar la centrifugación simplemente para eliminar partículas grandes o agregados de una solución de moléculas. Para ello es suficiente usar un rotor de ángulo fijo y a una velocidad baja. Esto se suele hacer por ejemplo para purificar proteínas.
La centrifugación también puede utilizarse para separar distintos tipos de partículas o moléculas grandes unas de otras. En este caso la variante de la técnica que se aplica es la “centrifugación en gradiente de sacarosa”, en la que se coloca la muestra en un tubo con una disolución de sacarosa con distintas concentraciones a distintas alturas del tubo, estando la más concentrada abajo. El gradiente se consigue con un aparato especial que va dejando caer en el tubo cantidades de sacarosa a distintas concentraciones.
La muestra la colocamos en la parte superior del gradiente y en principio quedará flotando por diferencia de densidad. Luego centrifugamos con un “rotor oscilante” o “basculante”. Aquí el tubo cuelga del rotor por una bisagra, y queda en posición horizontal debido a la fuerza centrífuga al rotar. Esta fuerza situará en bandas a los distintos componentes de la muestra según sus densidades en las distintas concentraciones de sacarosa.
Finalmente separamos estas bandas perforando el fondo del tubo (que suele ser de plástico) con una jeringuilla y tomamos cada una de las fracciones.
Ésta es una técnica preparativa porque recuperamos el componente de la muestra a purificar. Si lo que queremos separar son más proteínas debemos recurrir a la ultracentrifugación.
La técnica de sacarosa puede usarse también en ultracentrifugación , que puede usarse de modo analítico. Con las proteínas la analítica nos permite averiguar los coeficientes de sedimentación de las proteínas y así caracterizarlas según este parámetro.
TEMA 4 y TEMA 5
TÉCNICAS DE AISAMIENTO, PURIFICACIÓN Y CARACTERIZACIÓN DE BIOMOLÉCULAS
Metodos o técnicas de cromatografía:
La cromatografía implica el paso de una solución a través de un medio que presenta una adsorción relativa para los distintos solutos. Puede desarrollarse en papel, capa fina o columna. Las dos primeras no son muy utilizadas en la actualidad, por ello veremos las de columna.
Las columnas suelen ser de vidrio y se rellenan con un material que puede adsorber moléculas de modo selectivo debido a alguna diferencia en su estructura química. Este material se introduce suspendido en una disolución tampón adecuada. A continuación, la muestra la colocamos en la parte superior de la columna, y vamos añadiendo solución tampón lentamente, la cual va pasando lentamente a lo largo de la columna, y recogemos por la parte inferior de la columna fracciones del tampón que salen.
Este proceso es la “ELUCIÓN”. Lo que sale de la columna es el “eluído”, y la disolución tampón que va atravesando la columna es el “eluyente”. Las moléculas que no se adsorben al material con el que rellenamos la columna o que lo hacen débilmente son las primeras en salir, y las recogemos en las primeras fracciones. Las moléculas que se adsorben más fuertemente tardan más en eluír y además en algunas ocasiones incluso es necesario cambiar la composición del tampón de elución para que estas moléculas se suelten y las podamos recoger en la fracción.
Tanto el relleno como la disolución tampón se seleccionan dependiendo de la base que desee usarse para la separación.
Según cual sea el fundamento de la separación, distinguimos 3 grandes tipos de cromatografía en columna:
-
de intercambio iónico
-
de afinidad
-
de exclusión molecular
• también estudiaremos : - HPLC
- cromatografía de gases
Las cromatografías de intercambio iónico
se usan para separar moléculas de acuerdo con sus cargas eléctricas. Aquí el material de relleno de la columna es una resina de intercambio iónico. Estas son polianiones o policationes. Supongamos que separamos 3 clases de moléculas de una mezcla: una con carga negativa, otra con carga positiva débil y otra con positiva fuerte. La resina que debemos usar en este caso es una aniónica, que lleva grupos cargados negativamente por lo que las moléculas de la mezcla que están cargadas negativamente pasarán a través de la columna sin adsorberse y se recogerán en las fracciones iniciales. Las moléculas con carga positiva serán ligadas por la resina y se unirán más fuertemente con las de mayor carga positiva. Para separarlas de la columna y recogerlas en el eluído usamos un tampón con un gradiente de NaCl ya que estas disoluciones salinas rompen las interacciones electrostáticas. Vamos metiendo aumentando gradualmente la concentración de NaCl en el tampón de elución. A una pequeña concentración recogemos en el eluído las partículas de carga positiva débil y a mayor concentración de sal las de cargas positiva fuertes.
Cromatografía de afinidad:
es más específica, se basa en que muchas proteínas interaccionan fuertemente con otras moléculas como por ejemplo sucede con las encimas y análogos de sustratos, o como sucede con los antígenos y anticuerpos. Las moléculas adecuadas (análogo del sustrato o el anticuerpo) se unen covalentemente al material con el que se rellena la columna y van a actuar a modo de anzuelos moleculares para pescar a la proteína adecuada. El resto de las moléculas que no se unen al anzuelo simplemente pasan a través de la columna y salen en las fracciones iniciales.
Para eluír las moléculas que se han unido al anzuelo lo que se hace es eluír con una sal tampón que mantenga moléculas anzuelo libres o bien algún otro reactivo que pueda romper la unión entre la proteína o análogo del sustrato o anticuerpo.
Cromatografía de exclusión molecular:
se denomina también de filtración en gel, y en este caso las moléculas las vamos a separar en función de su tamaño y no de sus propiedades químicas. En este caso la columna se rellena con esferas de un gel poroso. Es muy empleado el “sephadox”, un polímero ramificado de la glucosa. Se dispone de sephadox de distintas porosidades y seleccionamos aquella según el tamaño de las moléculas que queremos purificar de modo que las moléculas más pequeñas de la mezcla pueden penetrar en las esferas mientras que las más grandes no pueden hacerlo. La mezcla se aplica en la parte superior de la columna y se va eluyendo con un disolución tampón. Las moléculas más grandes se mueven más rápido ya que no pueden penetrar en las esferas del gel sino que se cuelan en los intersticios entre ellas, entonces estas saldrán más rápidamente en las primeras fracciones mientras que las moléculas pequeñas pueden entrar en las esferas y tardan más en salir, eluyéndose consecuentemente en orden decreciente de pesos moleculares.
La cromatografía en gel también sirve para determinar pesos moleculares ya que existe una relación lineal entre el logaritmo del peso molecular de las proteínas globulares y el volumen de elución en un rango de pesos moleculares que depende del tipo de gel usado.
Hacemos pasar una serie de proteínas de peso molecular conocido (patrones) y medimos en qué volumen recogemos estas y representamos en unos ejes coordinados el logaritmo del peso molecular frente al volumen de elución (Ve). Observamos una línea de puntos recta. Así podemos calcular el peso molecular a partir del volumen de elución con la gráfica.
Para construir una recta de calibrado de este tipo necesitamos medir una serie de parámetros de la columna que son :
• Vt o volumen total de la columna. Es el volumen que ocupa el gel más el volumen de los intersticios. Se determina haciendo pasar una molécula con bajo peso molecular como NaCl y midiendo el volumen en que la recogemos.
• Vo o volumen de exclusión. Es el volumen de los huecos que hay entre las partículas de gel. Para calcularlo hacemos pasar a través de la columna una molécula de peso molecular alto, generalmente azul de dextramo.
• Ve o volumen de elución, que es el volumen en el cual recogemos cada una de las fracciones que recuperamos de las proteínas.
En lugar del Ve podemos usar el Kav , que es el coeficiente entre la fase líquida y el gel:
Cromatografía HPLC
( High Performance Liquid Chromatography o Cromatografía líquida de alta eficacia):
Las cromatografías estudiadas hasta ahora eran lentas, ya que solo se aplicaba una presión hidrostática para que los líquidos se colasen por la columna. Por ello, la elución suele tardar varias horas. En este tiempo las moléculas más frágiles se pueden deteriorar. Otro problema con que nos encontramos es que al ser un proceso lento, la muestra tiende a difundir (esparcirse) a medida que baja a lo largo de la columna. Cuanto más dure la separación peor será la resolución.
La HPCL resuelve estos problemas ya que usa unas presiones entre 5.000 y 10.000 psi para hacer que los líquidos atraviesen más rápidamente la columna, con lo que las separaciones que tardaban horas se reducen a minutos y la resolución es más alta debido a que no hay tanta difusión.
Para poder hacer esto fue necesario desarrollar resinas no compresibles con las que rellenar la columna, y además el cambio de columnas de vidrio por unas metálicas, más resistentes para soportar las presiones.
Este tipo de cromatografía HPLC también puede ser usado como cromatografía de intercambio iónico, de afinidad...
Cromatografía de gases:
Hasta ahora el eluyente era un líquido. Aquí es un gas. Esta cromatografía tiene lugar con la columna rellenada de un sólido inerte que está finamente dividido de materiales muy diversos como tierra diatomeas, cromosol. Este material se empapa de un líquido no volátil como un aceite de silicona que constituye la “fase estacionaria”.
El eluyente es un gas inerte como helio, argón o nitrógeno que se hace fluir a velocidad constante a través de la columna.
La muestra debe ser volátil ( para las moléculas no volátiles como aminoácidos, se hacen reacciones químicas que las transformen en derivados volátiles) que se introducen en la columna disueltos con un disolvente orgánico que se evaporan al introducirlos a alta temperatura por calentamiento.
Los productos volátiles se van separando a lo largo de la columna según su reparto entre las dos fases, la estacionaria y la móvil (gas o “gas portador”). Como la muestra se volatiliza a alta temperatura, y es a la que se produce la operación, las columnas están metidas en un horno.
Los productos se van cuantificando a la salida de la columna mediante detectores (muy variados) que están acoplados a registradores que nos van a dar un registro o cromatograma en el que cada componente que hemos separado se va a corresponder con un pico que sale cada cierto tiempo de retención que nos permite identificar a qué molécula corresponde cada tipo, siempre por comparación de patrones conocidos.
El área de cada pico nos permite cuantificar el compuesto que hay en la muestra. Esta es una técnica analítica, cuantificamos y cualificamos. No es una técnica preparativa porque recuperamos cantidades mínimas.
Electroforesis:
Son aquellas en las que aplicamos un campo eléctrico a una solución con moléculas del soluto con carga positiva. Estas se desplazan hacia el cátodo y las de carga negativa hacia el ánodo. A este desplazamiento se le denomina electroforesis.
La velocidad de las moléculas en la electroforesis depende de 2 factores:
En primer lugar, guiando el movimiento está la fuerza producida por el campo sobre la partícula que se corresponde por donde “q” es la carga de la molécula en Culombios, y es la carga del campo eléctrico en . Pero resistiendo al movimiento está la fuerza de fricción que ejerce el entorno sobre la partícula que es siendo “v” la velocidad de la partícula y “F” su coeficiente de fricción, que depende del tamaño y forma de las partículas (moléculas) de modo que las grandes o asimétricas presentan coeficientes de fricción mayores que las pequeñas.
Cuando se activa el campo eléctrico, las moléculas se aceleran hasta alcanzar una velocidad en la que ambas fuerzas se igualan, siendo
A continuación se mueve constantemente a esa velocidad. Esta igualdad también la podemos expresar así: o “movilidad electroforética”, y es la velocidad de movimiento por unidad de fuerza de campo. Consecuentemente, la movilidad de una molécula es la electroforesis, y depende de su carga y dimensiones moleculares.
Aunque la electroforesis se puede llevar a cabo en disolución, normalmente se usa algún soporte. Las dos más corrientes son el papel y el gel.
La de papel se usa para separar moléculas cargadas pequeñas. Se hace que la tira de papel de filtro se extienda humedecida en un tampón entre dos cámaras que son las que contienen los dos electrodos. En el centro del papel se coloca una gota de la muestra a analizar y se conecta el campo eléctrico. Al cabo de una hora, cuando las moléculas se han separado, se seca el papel y se tiñe con un colorante que nos deja ver las moléculas que estamos separando. Cada molécula la identificamos como una mancha, y se habrá desplazado hacia el ánodo o cátodo según su carga, y una determinada distancia que depende de su carga y dimensiones.
Al igual que en la cromatografía cada componente lo identificamos comparándolo con factores conocidos.
Actualmente se usa más la electroforesis en gel, que se emplea para separar proteínas y ácidos nucleicos. Se coloca un gel que contenga la disolución tampón adecuada entre dos placas de cristal. Estos geles son láminas finas de tan solo uno milímetros de espesor. Los geles suelen ser de poliacrilamida o agrarosa. La primera es para separar proteínas y ácidos nucleicos de bajo peso molecular. La agarosa para separar ácidos nucleicos de mayor tamaño. El gel se coloca entre los compartimentos que tienen los electrodos.
La muestra se aplica en la parte superior del gel, en los “pocillos” (hendiduras) y se le añade glicerol y un colorante. El primero es para que la muestra sea densa y se quede en el pocillo y no difunda por el tampón que hay en receptáculo superior. El segundo (colorante) nos sirve para seguir el proceso de la electroforesis. Cuando el colorante llega abajo, desconectamos, y aquí se supone que las moléculas ya están separadas. Con esto realizado retiramos el gel y lo teñimos con un colorante que nos permita visualizar las moléculas que estamos separando, que las veremos como bandas estrechas.
Si la electroforesis se realiza en gel, la movilidad es menor de la que presentan las moléculas en disolución por el efecto de tamizado molecular que ejerce el soporte, que es más fuerte cuanto más concentrado esté el gel.
Para moléculas cuya carga es proporcional a su longitud, como el DNA, que presenta una unidad de carga en cada residuo, presentan una movilidad libre casi independiente de su tamaño. Entonces, en la electroforesis en gel la movilidad de estas moléculas viene determinada por el efecto del tamizado molecular del gel.
Así, mediante electroforesis en gel podemos separar las moléculas casi exclusivamente por su tamaño.
Representamos el logaritmo del peso molecular frente a la distancia recorrida, y obtenemos unos puntos que se ajustan a una línea recta.
Técnicas de Diálisis y Ultracentrifugación
La diálisis y la ultracentrifugación sirven para concentrar y purificar biopolímeros. Se basan en la utilización de membranas semipermeables que permiten el paso de moléculas pequeñas pero no de proteínas o otras macromoléculas.
La diálisis se usa normalmente para eliminar moléculas pequeñas contaminantes o para cambiar las condiciones de una solución tampón.
Habitualmente se coloca la muestra (disolución de una proteína, por ejemplo) en una bolsa cerrada pero con las paredes de membranas semipermeables y se sumerge en un volumen mayor de una disolución tampón dentro de un recipiente. El tampón se somete a una agitación mediante un agitador magnético situado debajo de la muestra en la bolsa.
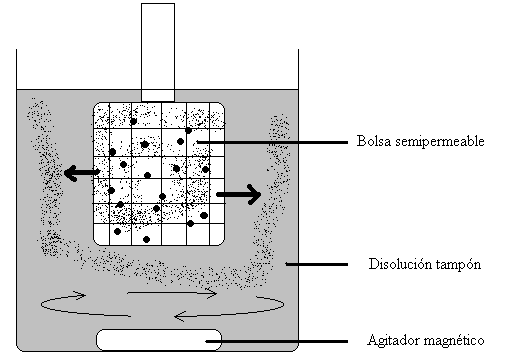
Las moléculas pequeñas “contaminadoras” saldrán de esta bolsa hacia la disolución tampón, y esta tenderá a reemplazar el líquido disolvente en el que tenemos la muestra. Si reemplazamos el tampón varias veces, al final tendremos la proteína disuelta en la disolución tampón exterior y habremos eliminado otras moléculas pequeñas que al principio la acompañaban.
La ultracentrifugación se utiliza para concentrar disoluciones de macromoléculas. También se usan membranas semipermeables, pero se aplica presión para hacer salir el disolvente y las moléculas pequeñas. Hay una gran diversidad de métodos. El más sencillo consta de un recipiente con una membrana semipermeable al que se le aplica una presión con gas como se indica en el dibujo:
La diálisis purifica, y la ultracentrifugación concentra. Otra técnica es la diafiltración, una combinación de las anteriores, donde se concentra y se eliminan contaminantes al mismo tiempo.
Primero se concentra la muestra por ultracentrifugación y se le añade la disolución tampón (gran volumen) y la volvemos a concentrar hasta que queda un pequeño volumen, y así sucesivas veces.
Técnicas de Radiactividad
Los isótopos radiactivos se empezaron a utilizar después de la segunda guerra mundial, y presentaron un gran avance ya que amplían en varios órdenes de magnitud la sensibilidad con la que detectamos una especie química. Los análisis tradicionales permiten detectar moléculas en cantidades mínimas de “nanomoles” (moles), mientras que con estas técnicas llegamos a una sensibilidad de “fentomoles” (moles).
Los compuestos marcados radiactivamente son los “trazadores”, ya que se pueden seguir las transformaciones que sufren en presencia de un exceso de material no radiactivo.
En bioquímica se usan dos isótopos, los radiactivos y los estables. En el caso del hidrógeno, tenemos el tritio (), radiactivo, y el deuterio (), estable.
La utilidad de los isótopos estables la resumimos en:
- incorporando un isótopo estable aumentamos la densidad de un material, lo que nos proporciona un método físico para separar los compuestos marcados de los no marcados.
-
Los compuestos marcados con isótopos estables, en especial el , se usan mucho en experimentos de resonancia magnética nuclear para el estudio de la estructura molecular y de los mecanismos de reacción.
- Como trazadores, cuando no se dispone de los radiactivos adecuados de ese elemento, como es el caso del y , que no poseen radiactivos
De entre los isótopos radiactivos, en bioquímica se usan los que emiten radiaciones y , pero no con emisiones . - Una es un electrón emitido
- Una es una radiación electromagnética (fotón con alta energía)
Las emisiones se usan sobre todo en inmunología ya que existen isótopos del yodo que son emisores y es fácil unir estos isótopos a anticuerpos. Salvo esta excepción, en bioquímica, los más usados son los emisores de radiaciones .
La radiactividad es un proceso cinético de primer orden: el número de desintegraciones que se producen en un determinado intervalo de tiempo depende solamente del número de isótopos radiactivos presentes.
Este fenómeno da lugar a la ley de desintegración radiactiva que tiene esta expresión:
, donde: - es el número de átomos radiactivos en el “t” = 0
- N es en nº de átomos que quedan en el tiempo “t”
- es la constante de desintegración
La constante de desintegración es característica para cada isótopo, y está relacionada con otro parámetro que se emplea más, que se llama “hemivida” o , que es el tiempo requerido para que se desintegren la mitad de los núcleos de una muestra, y es igual a:
o La hemivida es característica de cada isótopo.
La unidad básica de desintegración es el “Curio” (Ci). 1 Ci es la cantidad de radiactividad equivalente a la que existe en un gramo de radio, es decir, desintegraciones por minuto. En bioquímica se trabaja con cantidades de microcurios ().
Los detectores de radiactividad de que disponemos no presentan una eficacia del 100%, por lo que la radiactividad se expresa en unas unidades relativas que son las cuentas por minuto (c.p.m.), el número de desintegraciones que detecta el aparato en un minuto (menos que las reales).
Por ejemplo, un contador de radiactividad con una eficacia del 50% para una muestra de 0,1 nos daría un “contage” de c.p.m.
Para medir la radiactividad disponemos de contadores “GEIGER” y de contadores “de centelleo”, más empleados.
Los contadores geiger se basan en el fenómeno de la ionización que producen las emisiones radiactivas sobre un medio gaseoso. Estos se usan principalmente para saber, de modo relativo, si hay o no radiactividad en una muestra, y para cuantificarla pero de modo no muy aproximado.
Los contadores de centelmo nos ofrecen una mayor precisión. Se basan en los procesos de excitación. Aquí la muestra se disuelve o suspende en un disolvente orgánico que incluye uno o dos compuestos fluorescentes. Una partícula emitida por la muestra tiene una elevada probabilidad de contactar con una molécula del disolvente. Al ocurrir esto, la molécula del disolvente es excitada, haciendo que uno de sus electrones pase a un orbital de energía superior. Cuando este electrón vuelve a su estado inicial se emite un fotón de luz. Este fotón es absorbido por una molécula de flúor que a su vez se excita.
La fluorescencia implica la absorción de luz a una determinada energía seguida de la emisión de luz a una energía inferior o longitud de onda mayor .
Un fotomultiplicador detecta este pequeño destelleo de luz y lo convierte en una señal eléctrica que, que es la que se “cuenta”.
Los isótopos emisores presentan un espectro electromagnético característico cada uno. Estas diferencias se usan en los contadores de centelleo líquido para cuantificar simultáneamente dos isótopos de la misma muestra.
Las aplicaciones de los isótopos radiactivos en bioquímica son muy variadas. Se incluyen el estudio de rutas metabólicas con trazadores, estudios de cinética encimática con sustratos marcados, y la autoradiografía, que se basa en la capacidad de las emisiones radiactivas para impresionar placas fotográficas. Esta técnica combinada con la electroforesis nos permite, por ejemplo, secuenciar el DNA o hacer estudios de la expresión de los genes, entre muchas otras cosas.
Técnicas espectroscópicas
Tanto proteínas, como ácidos nucleicos o hidratos de carbono, son moléculas complejas que pueden absorber radiación en un amplio margen del espectro electromagnético. En la región infrarroja del espectro se estudia la conformación de las moléculas proteicas.
La región visible se usa para la medida de la absorbancia de las soluciones coloreadas, y esto es conocido como “colorimetría”.
En la zona visible la mayoría de los biopolímeros no absorben luz. Aunque algunas proteínas se ven coloreadas, ello es debido a los grupos prostéticos o iones, como el cobre que forman parte del polipéptido. El color rojo de la sangre se debe al grupo “hemo” de la hemoglobina.
Para poder medir las proteínas o ácidos nucleicos por colorimetría, hay que transformarlos en compuestos coloreados mediante reacciones químicas específicas.
En la región ultravioleta absorben luz los ácidos nucleicos y las proteínas, midiéndose respectivamente a 260 y 280 nm. En realidad se mide en las cadenas laterales aromáticas de los aminoácidos Phe, Tyr y Trp.
Las medidas se absorbancia de luz ultravioleta y visible se hacen en unos aparatos llamados “espectrofotómetros”.
La muestra se coloca en una cubeta sobre la que incide una luz monocromática (con una sola longitud de onda) que se consigue con un monocromador. Parte de esta luz incidente va a ser absorbida por la muestra y otra va a atravesarla.
La que la atraviesa es la luz “i”, y es detectada y registrada.
La absorbancia a la longitud de onda “” se define como el logaritmo del cociente entre Io e I: y se relaciona con la concentración mediante la ley de LAMBERT - BEER que dice que la absorbancia es igual a Epsilon por la concentración y por el grosor de la cubeta:
es el coeficiente de extinción, característico para cada sustancia. Sus unidades dependen de las unidades de la concentración que se emplean ya que la absorbancia no tiene unidades.
El grosor de la cubeta suele ser de un centímetro, por lo tanto si la concentración la expresamos como molaridad, las unidades de serían
Esta ley nos permite calcular concentraciones de una sustancia a partir de su absorbancia.
TEMA 6
HIDRATOS DE CARBONO, GLÚCIDOS
Concepto, clasificación e importancia biológica
Los carbohidratos, puramente, son aldehídos o cetonas con múltiples grupos hidroxilo (polihidroxialdehídos o polihidroxiacetonas). Constituyen la mayor parte de la materia orgánica de la tierra por sus variadas funciones, de entre las que destacamos:
-
Actúan como almacenes de energía, combustibles e intermediarios metabólicos
-
Forman parte de los ácidos nucleicos ADN y ARN (desoxirribosa y ribosa)
-
Son elementos estructurales en las paredes celulares de plantas y bacterias, y en el exoesqueleto de artrópodos
-
Desempeñan funciones importantes unidos a proteínas y lípidos como la de reconocimiento intercelular
La clasificación más sencilla depende del número de unidades de azúcar de que consta. Así vamos a tener - las “osas” o monosacáridos
- oligosacáridos ( aprox. Entre 2 y 12 unidades )
- polisacáridos ( más de 13 )
A los oligosacáridos y polisacáridos se les llama también “ósidos” , que pueden ser:
- holósidos (formados solo por monosacáridos)
- heterósidos ( “ tambíen por una fracción no glucídica (glucagona))
Los monosacáridos
Muchos de ellos tienen como fórmula empírica , y por ello se les llamó hidratos de carbono. Los clasificaremos según dos criterios:
-
Según el número de átomos de C ( triosas, tetrosas, pentosas, hexosas...)
-
Según su grupo funcional : - Aldehído aldosas
- Cetona cetosas
Lo habitual es combinar los dos criterios, y así tenemos aldopentosas, cetotetrosas...
Los monosacáridos más sencillos tienen 3 átomos de C (aldotrioas y cetotrioas). En el caso de las aldotriosas vamos a tener 2, debido a que la aldotriosa el el gliceraldehído , y éste posee un C asimétrico (C) (el carbono 2). Distinguiremos el D-gliceraldehído y el L-... dependiendo de si el OH del Cestá a la derecha o izquierda respectivamente.
CHO
ø
H _ "C _ OH D-gliceraldehído
ø
CH2OH
Las formas D y L son “enantiómeros” porque son estereoisómeros ópticos (son imágenes especulaes uno con respecto al otro) y además son no superponibles.
Si añadimos un nuevo átomo de C, en lugar de una aldotriosa tenemos una aldotetrosa. A partir del D-gliceraldehído tenemos 2 tetrosas, y a partir del “L” otras 2, ya que este nuevo carbono añadido es asimétrico y por tanto podrá presentarse con dos configuraciones distintas, una con el OH hacia la derecha y otra hacia la izquierda.
Estas tetrosas formadas son la D-eritrosa y la D-treosa, que pertenecen a la serie “D” ya que se forman a partir del D-gliceraldehído, el cual tiene el OH a la derecha del carbono más alejado del grupo funcional (aldehído).
La D-eritrosa y la D-treosa son entre sí “diastereoisómeros” porque no son imágenes especulares.
Si añadimos un nuevo átomo de C, ésta molécula también podrá presentar las dos conformaciones al ser aquel asimétrico.
La D-eritrosa nos dará lugar a dos pentosas, al igual que la D-treosa. Son importantes la D-ribosa y la D-Xilosa.
Si seguimos este proceso de adición de 1 C, obtendremos de cada aldopentosa 2 aldohexosas. Las más importantes son la D-glucosa y la D-galactosa, que son “epímeros” entre sí ya que solo se diferencian en la configuración de un C, el , en el que la glucosa tiene su OH a la derecha.
En general, cuando una molécula no contiene planos de simetría, el número de estereoisómeros posibles es , siendo “n” el número de carbonos asimétricos.
En el caso de 1 hexosa, tenemos 4 C, y por tanto estereoisómeros posibles, 8 de la forma “D” y otros 8 de la forma “L”.
En el caso de las cetosas todo esto es diferente, aquí el número posible de estereoisómeros es menor ya que la cetosa más pequeña es la dihidroxiacetona, y esta nó presenta carbonos asimétricos y solo puede encontrarse con una única conformación, no como en el caso del gliceraldehído.
CHOH
ø
C = O Molécula de dihidroxiacetona
ø
CHOH
Añadiendo un C como hicimos anteriormente con la otra serie, obtenemos una cetotetrosa, que es la “eritrulosa”.
Las cetosas de 4 C tienen el asimétrico, por lo que podrá existir como 2 estereoisómeros, la D-eritrulosa y la L-eritrulosa.
Como es una cetosa, su nombre termina en “ulosa”. La terminaciones de los aldehídos es“osa” (eritrulosa).
Si a la D-eritrulosa le añadimos un átomo de C con su correspondiente OH, obtendremos dos cetopentosas de la serie “D”, la D-ribulosa y la D-xilulosa. Sus configuraciones (D o L) nos viene indicado (como antes) por el C más alejado del grupo funcional. De todos estos, es importante la D-fructosa.
Tanto las pentosas como las hexosas se pueden ciclar en anillos. En el caso de una aldohexosa, como la glucosa, reaccionan el grupo carbonilo () y el grupo OH del , formando un “hemiacetal intramolecular”. Lo que resulta es un anillo de 6 eslabones, llamdado “piranósico”.
Una glucosa en esta forma cíclica, también se llama glucopiranosa, y es la forma que predomina de esta en disolución.
Cuando una aldohexosa se cicla, aparece un nuevo C , ya que el ,aunque en la forma abierta no es asimétrico, al ciclarse se convierte en uno, y por tanto da lugar a que aparezcan dos nuevos estereoisómeros llamados “anómeros”.
Al se le denomina “anomérico”.
Estos dos nuevos anómeros se les llama y , que se van a diferenciar en que en la “proyección de Haworth” el anómero presenta el OH del hacia abajo, mientras que el anómero hacia arriba.
Los anillos, realmente forman un plano, y los sustituyentes de los carbonos (en los vértices del anillo) se sitúan hacia arriba y hacia abajo de este. Por ejemplo, en el caso de la glucosa, cuando el OH está hacia abajo tenemos la -D-glucopiranosa, y si está hacia arriba, entonces la -D-glucopiranosa.
En el caso de las cetosas como la D-fructosa, la reacción se produce entre el grupo ceto del
y el OH del . El enlace de este caso es un “hemicetal” intramolecular, y da lugar a un anillo de 5 elementos, que se denomina “furanósico”. En este caso también cuando se cicla la fructosa ( o cualquier cetosa) aparece un nuevo C “anomérico” que en este caso es el , y que posee sus respectivos enómeros y .
La fructosa también puede formar un anillo piranósico (de 6 lados), formando un hemiacetal intramolecular entre los carbonos 2 y 6, teniendo un anillo como el de la glucosa. De echo, la forma piranósica es la que predomina de forma libre, pero cuando está unida a algún sustituyente predomina la furanósica.
En disoluciones acuosas los monosacáridos presentan la denominada “mutarrotación”, que, por ejemplo, en el caso de la glucosa se produce la interconversión de la -D-glucopiranosa y la -D-glucopiranosa a través de la forma de cadena abierta en disolución, con lo que se alcanza un equilibrio en el cual tenemos del anómero y 2 del , representando la forma de cadena abierta menos del 1%.
La mutarrotación se detectó midiendo el poder rotatorio de disoluciones acuosas de monosacáridos, en las cuales este cambia con el paso del tiempo.
Aunque las proyecciones de Haworth son en un plano, en realidad estas moléculas no son planas, sino que adoptan dos tipos de conformación, la de “silla” y la de “bote”.
Los sustituyentes de los carbonos van a ser de 2 tipos, axiales y ecuatoriales. Los axiales son perpendiculares al plano central del anillo, y los ecuatoriales son paralelos a este.
Cuando los sustituyentes axiales son grupos distintos del H, se estorban estéricamente , y por tanto, de estas dos conformaciones, la más estable es la que tenga más hidrógenos en posición axial.
En el caso de la -D-glucopiranosa, la conformación más estable es la de silla, en la que todos los sustituyentes están ocupados por H.
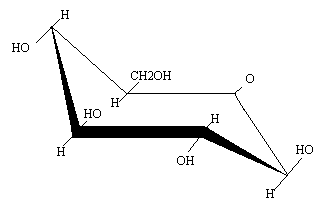
Los anillos furanósicos tampoco son planos. Aquí la conformación adoptada se denomina “de sobre” ya que tienen forma de sobre con la solapa abierta. 4 átomos del anillo son coplanares, y el 5º se situa en otro plano, formando “la solapa”.
Tomando como ejemplo la -D-ribosa, el átomo que se aleja puede ser el o el , llamándose respectivamente “´-endo” y “´-endo”.
La ribosa que forma parte del RNA se encuentra en la conformación ´-endo, mientras que la dexosirribosa del DNA en la ´-endo.
Estos anillos furanósicos son más flexibles que los piranósicos, y pueden convertirse uno en otro con mayor facilidad. Esto les da más preferencia para formar parte de los ácidos nucleicos, frente a las aldohexosas.
Determinación del enlace oxídico
Esta consiste en averiguar, en un monosacárido, cuales son los átomos implicados en enlace oxídico. Para ello se emplean dos métodos fundamentales.
El primero consiste en el tratamiento del monosacárido con un ácido periódico, que rompe los enlaces C-C de las cadenas de los monosacáridos, pero no cuando estos participan en el puente oxídico.
En los monosacáridos hay alcoholes (OH) primarios y secundarios. Por tratamiento con periódico, a partir de los primarios obtenemos “aldehído fórmico”, y a partir de los secundarios “ácido fórmico”.
Existe otro método que puede usarse complementariamente, que es la “metilación” del monosacárido. Los OH se pueden metilar por tratamiento de yoduro de metilo en presencia de plata o con sulfuro de metilo en medio alcalino. Así se nos metilan todos los OH excepto los del puente oxídico.
Si después de la metilación realizamos una hidrólisis ácida suave con HCl diluido, eliminamos el grupo metilo del C anomérico pero no los otros. A continuación identificamos este compuesto por cromatografía, y en función de los carbonos metilados sabremos donde estaba el puente oxídico.
TEMA 7
PROPIEDADES Y DERIVADOS DE LOS MONOSACÁRIDOS
Propiedades físicas y químicas de los monosacáridos
Los monosacáridos son - blancos y cristalinos,
- solubles en pero no en disolventes orgánicos,
- tienen sabor dulce ,
- presentan poder reductor, debido al grupo carbonilo (en el caso de las cetosas esto solo se manifiesta en medio alcalino).
- presentan isomería óptica por sus carbonos asimétricos.
Los monosacáridos se pueden identificar porque no absorben la luz ultravioleta, pero en la infrarroja presentan un espectro característico.
Se pueden identificar en luz visible por colorimetría, pero hay que transformarlos en compuestos coloreados previamente.
También se pueden identificar por cromatografía.
Reacciones y derivados de los monosacáridos
Oxidación de los monosacáridos:
Puede producirse de diversas formas según cual sea el agente oxidante. Usando, por ejemplo, la oxidación suave de una aldosa en presencia del ión y en medio alcalino, obtenemos el “ácido aldónico”. El “reactivo de Fehling” es el usado para la oxidación.
Como consecuencia de la oxidación se forma de color rojo que nos permite identificar si hay o no un monosacárido reductor.
Otro tipo de derivados por oxidación son los “ácidos urónicos”, que se forman por oxidación suave en el grupo alcohol primario () , que está en el en la glucosa. Se oxida a un grupo ácido, y el ácido correspondiente es el glucurónico (en el caso de la glucosa). Los ácidos urónicos no se encuentran en estado libre.
En los ácidos aldáricos se oxidan los dos átomos de C con alcoholes primarios. Es una oxidación enérgica, y afecta al C del grupo carbonilo y al del OH primario.
El de la glucosa se denomina “ácido glucárico” o “sacárico”.
Conjuntamente con los ácidos aldáricos y aldónicos aparecen uno compuestos que químicamente son lactonas que se forman mediante una reacción de esterificación intermolecular con liberación de una molécula de agua. Por tanto, reacciona el grupo ácido del con uno de los grupos alcohólicos de la aldosa.
Aparecen la o la gluconolactona, dependiendo de que OH actúa, llamándose los carbonos de después del , “,, y ”respectivamente.
Los ácidos aldónicos están en equilibrio con las lactonas en disolución.
Las lactonas son compuestos importantes porque actúan como intermediarios del metabolismo. Además, destaca una por su importancia biológica, que es la vitamina C o “ácido ascórbico”, que se forma a partir de la glucosa, y es una -lactona de configuración “L” y dextrógira.
Formación de heterósidos mediante enlaces glicosídicos
Los heterósidos se forman por unión de moléculas de un monosacárido con otras moléculas. La unión se forma entre el C anomérico de un monosacárido y un heteroátomo de la otra molécula, llamándose a esta unión “enlace glicosídico”.
Según cual sea el heteroátomo diferenciamos los:
-
O-heterósidos, en los que el heteroátomo es el oxígeno, y el enlace es O-glicosídico.
-
S-heterósidos, “ “ “ azufre “ “ S-glicosídico.
-
N-heterósidos,” “ “ nitrógeno “ “ N-glicosídico.
Los O-heterósidos (también llamados O-glucósidos) se forman por condensación del monosacárido con un hidroxilo alcohólico o fenólico que aportan el O. En esta unión también se libera una molécula de .
o glucosa o metilglucósido
aunque los glucósidos pueden ser o , no se interconvierten mediante mutarrotación en disolución y por eso se emplean para determinar si los monosacáridos son o .
Si este OH aporta otro monosacárido se forma un disacárido unido por un enlace O-glicosídico.
Los S-heterósidos se forman por combinación las aldosas con moléculas que llevan un grupo “tilo” (SH). Se forman “mercaptales”.
Solo las aldosas dan esta reacción, no las cetosas, que reaccionan en su estructura lineal.
Los N-heterósidos se forman por condensación del monosacárido con un grupo aminado. Por ejemplo, los nucleósidos son N-heterósidos.
Además, hay dos derivados amino de los azúcares que tienen importancia biológica al aparecer en muchos polisacáridos. Uno de ellos es la glucosamina, en la que se sustituye el OH del por un grupo amino. En la glucosamina aparece en ocasiones acetilado el grupo amino (N-acetil-glucosamina). Esto pasa igual con la galactosa.
NOTA: La glucosamina y la galactosamina no son N-heterósidos, ya que no interviene el C anomérico en su formación.
Otros derivados de los azúcares son los ésteres fosfato, que se forman por combinación del ácido ortofosfórico con cualquiera de los grupos OH del monosacárido (anomérico u otro cualquiera).
Si el carbono que reacciona es el anomérico, hablamos de los ácidos osil-fosfóricos.
En el caso de la glucosa, el ácido D-glucosil-1-fosfórico es la glucosa-1-fosfato.
Existen monosacáridos importantes unidos a varios grupos fosfato. Diferenciaremos si estos fosfatos están sobre el mismo C o sobre otros. Por ejemplo, la fructosa 1,6 -difosfato ( que se recomienda llamar fructosa 1,6 -bisfosfato ) posee 2 fosfatos sobre 2 átomos de C distintos.
Las moléculas de ácido fosfórico pueden unirse unas con otras, y este conjunto a un C del monosacárido. En este caso el prefijo sería “di,tri...” como en el caso del ATP
Otros derivados importantes en biología son los “desoxiazúcares”, en los que se pierde un grupo OH . Por ejemplo, en la 2-desoxirribosa tenemos 1 H en lugar de un OH (en la ribosa). La desoxirribosa forma parte del ADN.
Otros derivados importantes en biología son los “alditoles” o azúcares-alcoholes, que son derivados por reducción del grupo carbonilo (aldehído o cetona) de los monosacáridos.
Los más importantes de la naturaleza son el D-manitol y el D-sorbitol.
Por ejemplo, de la glucosa obtenemos el sorbitol (o glucitol).
De las cetosas generamos un nuevo C, y así, por reducción de la D-fructosa, el OH que aparece, si está a la derecha, es el D-sorbitol, y si está a la izquierda, el D-manitol. Estos dos son empímeros.
Reacciones para identificar monosacáridos
En medio alcalino, los monosacáridos sufren 2 reacciones:
-
Interconversión aldosa - cetosa (epimerización)
-
Deshidratación
La deshidratación se produce por acción de los ácidos y además en un medio ácido fuerte. A partir de los OH se van liberando y se forma un compuesto cíclico llamado “furfural”. 1 hexosa hidroximetil-furfural
Tanto uno como otro se condensan con fenoles o compuestos nitrogenados cíclicos, para dar lugar a compuestos coloreados.
Por tanto, estas reacciones se usan para la identificación de los monosacáridos.
La “reacción de Molish” consiste en la combinación de -naftol con los monosacáridos dando un compuesto de color violeta indicativo de la presencia de aquellos.
TEMA 8
OLIGOSACÁRIDOS
Los oligosacáridos se forman por la unión de monosacáridos mediante enlaces O-glicosídicos. Por tanto, uno de los C que participan en el enlace debe ser anomérico.
Aunque los oligosacáridos contienen entre 2 y 12 monosacáridos aproximadamente, los más importantes y abundantes son los disacáridos (2 monosacáridos).
Clasificación e importancia biológica. Oligosacáridos más importantes
Los disacáridos se nombran y clasifican en función de su carácter reductor. Hay disacáridos reductores y no reductores. Esto depende de su grupo carbonilo, que es el que le confiere dicho poder reductor. Los oligosacáridos en los que los dos grupos carbonilo participan en el enlace no presentan poder reductor, debido a que no queda libre ningún grupo carbonilo en la molécula. En los disacáridos que solo intervenga un grupo carbonilo se dará poder reductor al quedar uno libre.
Los oligosacáridos se nombran en función de su carácter reductor, de los monosacáridos que lo formen, y de sus enlaces.
Por ejemplo, la maltosa, deriva de la unión de 2 D-glucosas (Glc). Una Glc reacciona con su carbono anomérico y la otra reacciona con el OH de su . La maltosa, por todo esto, se nombraría así: -D-glucopiranosil (14) -D-glucopiranosa (nomenclatura IUPAC)
La terminación “osa” nos indica que es reductor.
Hay otra nomenclatura “abreviada” en la que los monosacáridos se designan por 3 letras, y nuestro ejemplo quedaría de la forma: Glc (14) Glc (reductor).
Un disacárido no reductor es la “sacarosa”, formada por la unión de 1 Glc + 1 Fru (fructosa). En el enlace glicosídico intervienen los dos carbonos anoméricos. Exactamente la unión sería de:
-D-glucosa (con OH hacia abajo) + -D-fructosa
Normalmente la -D-fructosa se dibuja dada la vuelta para facilitar la representación del enlace.
Esta molécula se nombraría así : -D-glucopiranosil (12) -D-fructofuranósido, y abreviadamente: Glc (12) Fru.
Hasta ahora hemos visto enlaces , que forman un vértice hacia abajo, en el que el O forma su “punta”. En los el O hace el vértice hacia arriba.
Los más importantes son la “celobiosa” (de la celulosa) y la lactosa.
La celobiosa es la -D-glucopiranosil (14) -D-glucopiranosa o Glc (14) Glc.
La lactosa, presente en la leche, se forma por unión entre una D-galactosa y una D-glucosa, y se nombra -D-galactopiranosil (14) -D-glucopiranosa o Gal (14) Glc.
Para nombrar oligosacáridos mayores la regla sería la misma. Normalmente el orden es empezando de izquierda a derecha desde el extremo no reductor hasta el reductor (con el C anomérico libre).
Con relación a los trisacáridos, existen pocos importantes en biología. Destaca la “rafinosa”, presente en la remolacha, que resulta de la unión de 1galactosa + 1 sacarosa.
Gal (16) Glc (12) Fru
sacarosa
rafinosa
Dentro de los oligosacáridos vamos a ver también las oligosacarinas, que actúan como hormonas vegetales.
La primera que se descubrió es un heptaglucósido, 7 glucosas unidas mediante 5 uniones (16) y 2 uniones (13) de la siguiente manera:
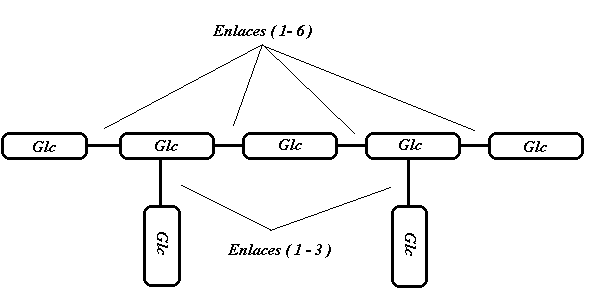
las uniones (13) son puntos de ramificación que son críticos, en el sentido de que si se modifican (cambiando su posición en la cadena), se altera su actividad biológica.
Identificación y análisis estructural
Para realizar el análisis estructural de los oligosacáridos se usan reacciones que hemos visto para los monosacáridos.
-
En primer lugar se analiza si es reductor o no reductor, usando la “racción de Fehling”.
-
En segundo lugar se hidrolizan e identifican lo monosacáridos que lo forman por cromatografía
-
Para saber el carácter o del enlace glicosídico se hidroliza con encimas específicas de estos o bien se observa la mutarrotación de los productos de hidrólisis. Si el enlace es el poder rotatorio va disminuyendo con el tiempo.
Para determinar los carbonos que están en el enlace glicosídico, en primer lugar reducimos el grupo carbonilo hasta un alcohol con boro-hidruro de sodio, un agente reductor (). Al reducirse también se rompe el puente oxídico.
A continuación el derivado reductor se metila a fondo (todos los OH que se puedan metilar) y a continuación se hidroliza.
Solo quedan sin metilar los grupos hidroxilo implicados en el enlace glicosídico.
Los derivados metilados se identifican por cromatografía.
Por último, la estructura se confirma por oxidación con periodato y análisis de los fragmentos resultantes.
Por ejemplo: Averigua la estructura de un disacárido que:
-
es positivo en la reacción de Fehling
-
por hidrólisis y cromatografía se sabe que está formado solo por Glc
-
por metilación exhaustiva, seguida de hidrólisis en medio ácido obtenemos 2,3,4,6-tetrametil-glucosa y 3,4,6-trimetil-glucosa
Como da positivo en la reacción de Fehling, posee un carbono anomérico libre y es reductor
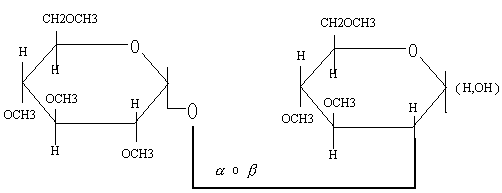
la única posibilidad de formación del enlace glicosídico es (12) ya que los demás no están en condiciones de formar enlace.
Determinantes antigénicos de los grupos sanguíneos
Son un grupo de oligosacáridos que están unidos a la superficie de los glóbulos blancos mediante proteínas de membrana o a través de lípidos. Quedan expuestos, y en algunos casos inducen la formación de anticuerpos y en otros no.
Las personas podemos producir anticuerpos frente a los oligosacáridos de los tipos A y B, pero no frente a los 0.
La diferencia entre los A y B, y el 0 está solo en una unidad de monosacárido complementario. El esqueleto básico es común.
TEMA 9
POLISACÁRIDOS
Concepto y clasificación
Los polisacáridos están formados por la unión de más de 12 monosacáridos por enlaces O-glicosídicos. Se suelen clasificar como:
-
homopolisacáridos: formados por el mismo monosacárido
-
heteropolisacáridos: “ por varios tipos de monosacáridos diferentes
Otra clasificación es la de polisacáridos
-
simples : formados por monosacáridos simples (Glc,Gal...)
-
derivados: “ por derivados de monosacáridos (acetilados, azúcares ácidos...)
Glucanos más abundantes
La celulosa es la molécula de origen natural más abundante que existe en la naturaleza. Esto se debe a que la celulosa forma parte de las paredes celulares de los vegetales, y estos forman una gran biomasa. Su función es pues, estructural.
Vista al microscopio electrónico, la celulosa está formada por fibras que están, a su vez, formadas por un conjunto de cadenas de glucosa no ramificadas y dispuestas en paralelo unas a otras.
Todas las unidades son -D-Glc unidas por enlaces glicosídicos (14). Es un “homopolisacárido simple”. La unidad repetitiva es el disacárido “celobiosa”.
La celulosa se organiza en cadenas lineales, y estas en fibras, por el establecimiento de puentes de H.
Dentro de la propia cadena de celulosa se pueden establecer puentes de H que mantienen esta estructura lineal, pero también entre cadenas paralelas de celulosa.
Esta estructura es ideal para su función biológica. Su elevada capacidad de formar puentes de H le da firmeza y es responsable de la insolubilidad en agua de este polisacárido.
En las paredes celulares de las plantas, estas fibras de celulosa se refuerzan por la presencia de otras sustancias que actúan a modo de cemento, como son la “hemicelulosa”, “pectina”, y “lignina”.
La hemicelulosa es un polisacárido formado por D-xilosa. La pectina está formada por el ácido D-galacturónico, y la lignina es una sustancia más compleja (polifenólica).
Todas ellas contribuyen a la rigidez de la pared.
La celulosa no puede ser digerida por los animales superiores excepto en el caso de los rumiantes. Además de estos últimos, también pueden aprovecharla diversos microorganismos, insectos, caracoles... A todos ellos se les denomina “celulolíticos”.
La quitina es otro polisacárido relacionado con la celulosa. Está formada por la unión de moléculas de N-acetil-glucosamina, unidas por enlaces (14).
Al igual que sucedía con la celulosa, como los enlaces son (14), para que el enlace glicosídico se pueda formar, una molécula tiene que estar invertida con respecto a la otra.
La unidad repetida en este caso es el disacárido “quitobiosa”.
Como en la celulosa, en la quitina se pueden formar puentes de H, y por tanto es un polisacárido que también cumple una función estructural. Es el principal componente del exoesqueleto de artrópodos y también de las paredes celulares de microorganismos como levaduras y hongos.
Polisacáridos con función de almacenamiento
El almidón es un polisacárido que está presente en los vegetales de modo exclusivo. Está formado por la unión de moléculas de glucosa (-D-Glc), y nos lo encontramos en forma de gránulos citoplasmáticos o también dentro de los plastos de los vegetales.
Los enlaces pueden ser de 2 tipos porque el almidón está formado por 2 componentes, la “amilosa” y la “amilopectina”.
En la amilosa, los enlaces son todos (14), mientras que en la amilopectina, son también (14), pero a intervalos regulares nos encontramos con enlaces (16). Es decir, la amilosa está formada por cadenas lineales, y la amilopectina por cadenas ramificadas, de modo que cada enlace (16) representa un punto de ramificación.
La estructura es estos dos componentes es la siguiente:
La amilosa es helicoidal, debido al establecimiento de puentes de H. En las cadenas de amilosa también se establecen puentes, pero de modo diferente a los de la glucosa.

La estructura secundaria de la amilopectina no se conoce con tanto detalle. Posee forma arborescente debida a las ramificaciones.
Cada molécula de celulosa presenta un extremo reductor y uno no reductor. Esto mismo pasa con la amilosa.
En el caso de la amilopectina, una molécula tiene un único extremo reductor pero muchos no reductores, debido a sus ramificaciones (tantos como ellas).
Otro polisacárido que también está relacionado funcional y estructuralmente con la amilopectina es el glucógeno, polisacárido de reserva del medio animal. Se encuentra en el hígado y en el músculo.
Su estructura es similar a la de la amilopectina, pero este está más ramificado. Los enlaces son (16), y se sitúan cada 8 unidades de glucosa en el glucógeno, y en la amilopectina cada 25 - 30. Ambos son digeribles por los animales superiores (con encimas “amilasa”).
Mucopolisacáridos ácidos o glicosaminoglicanos
Son importantes en el reino animal, y más en los vertebrados. La características generales de este tipo de polisacáridos son que:
están formados por aminoazúcares, normalmente por N-acetil-glucosamina y N-acetil-calactosamina, y de aquí sus nombres.
Suelen poseer grupos cargados negativamente (carbonato, sulfato).
No suelen existir en estado libre, sino asociados a proteínas, formando los proteoglicanos, o también a asociaciones de lípidos y proteínas.
Dentro de estos glicosaminoglicanos vamos a estudiar la estructura de tres fundamentalmente:
El “ácido hialurónico” lo encontramos en el tejido conectivo (en la sustancia basalque rellena los espacios extracelulares de éste), en el líquido senovial, humor vítreo y cordón umbilical.
Se trata de un polímero lineal no ramificado. La estructura que se repite es un disacárido formado por la unión del ácido D-glucurónico y de la N-acetil-glucosamina, mediante enlaces (13). Cada disacárido se une al siguiente por enlaces (14). Estos dos enlaces se alternan en la cadena.
Presentan una gran viscosidad, debida a la presencia de estos grupos ácidos que a PH fisiológico presentan carga negativa, están ionizados(), lo que favorece una hidratación excesiva, y la formación de puentes de H intercatenarios.
Otros dos glicosaminoglicanos también asociados con el tejido conectivo son el “sulfato de condroitina o condroitín sulfato” y “el queratán sulfato”. En ambos, el ptatrón de enlaces glicosídicos es alternante (13), (14) como en el caso del ác. hialurónico.
En su composición intervienen N-acil-azúcares y también llevan grupos cargados negativamente, pudiendo aparecer grupos sulfato ahora (). En el caso del condroitín , el disacárido está formado por el ác. D-Glucurónico y por la N-acetil-D-galactosamina. Aquí la N-acetil-gal está sulfatada en el .
La longitud de la cadena del sulfato de condroitina puede variar entre 15 y 150 unidades de disacárido, y la extensión de la cadena, así como el grado de sulfatación dependen de la edad del tejido y de algunas condiciones patológicas. En el caso del sulfato de queratán, el disacárido repetido está formado por la galactosa y la N-acetil-galactosamina, que se unen por enlaces (14) y (13) el intermediario. La galactosamina también está sulfatadoa en el . Aquí, a diferencia, interviene la galactosa simple, y no tenemos grupos carboxilato.
Aquí la longitud de cadena oscila entre 10 y 50 unidades de disacárido y es menor que la del sulfato de condroitina.
En resumen: Ácido hialurónico grupos carboxilato
Base común Heparán sulfato grupos carboxilato y sulfato
Queratán sulfato grupos sulfato
Otro tipo de mucopolisacáridos ácidos forman las paredes de las bacterias, y son un entramado estructural que rodea completamente a la célula.
Es una estructura continua (como una bolsa), constituída por cadenas de polisacáridos lagas y paralelas que están unidas entre sí transversalmente a ciertos intervalos por cadenas laterales que son péptidos cortos.
Los polisacáridos están formados por la unión de la N-acetil-D-glucosamina y el ácido N-acetil-murámico mediante enlaces (14). El componente ácido es un monosacárido complejo que lleva un radical en el , lugar donde va unida la cadena lateral polipeptídica corta que hace de unión d entre polisacáridos.
El péptido suele ser un tetrapéptido cuya composición varía entre las especies bacterianas.
A esta estructura continua que rodea a la célula se le denomina “péptidoglucano” o “mureína”. Hay algunos antibióticos como la penicilina, cuya acción consiste en inhibir una de las múltiples etapas de la síntesis encimática de los peptidoglucanos.
Enlaces azúcares - proteínas
Los dos más importantes que suelen aparecer en las proteínas son:
-
N-acetil glucosamina + cadena proteica: la unión se establece por el grupo amino de la “asparagina” y se llama una N-GLICOSILACIÓN.
-
Otro tipo de enlace es un O-glicosídico con la treonina o serina. O-GLICOSILACIÓN.
EJERCICIOS:
1.- Una muestra de 75 mg de celulosa se hidrolizó en medio ácido. En el hidrolizado se encontraron 75 mg de glucosa. ¿cuál es la pureza de la muestra de celulosa?
Como la unión Glc-Glc es un enlace glucosídico, se gana una molécula de en cada hidrólisis de 2 moléculas, y su peso hay que descontarlo.
El peso molecular de la glucosa es 180.
El peso “ de las glucosas unidas por enlace glicosídico es 180 - 18(peso del ) 180 - 18 = 162.
Entonces, la relación de pérdida de masa será de , que a su vez es el peso que aumentamos al hidrolizar, ya que al romper los enlaces les aportamos la molécula de agua que habían perdido al formarse el mismo.
Lo máximo que podríamos tener sería 75 + 1,1 , que sería el 100%
75 + 1,1 100%
75 x% x = 90,9%
2.- Por metilación exhaustiva de 32,4 gr de amilopectina seguida de hidrolización ácida se obtienen 10 de 2,3,4,6 tetrametil-glucosa.
¿Cuáles son los otros productos (derivados metilados) que espera obtener?
¿Cuánto de cada uno?
¿Qué tanto % de residuos de glucosa están unidos por enlaces (16)?
Si el peso molecular de la amilopectina es de , ¿Cuántas ramificaciones posee una molécula de amilopectina?
Amilopectina = glucosas con enlace (14) + ramificaciones periódicas con es.(16)
Hay cuatro tipos de Glc distintas: - La del C anomérico libre (una en cada molécula de amilopectina)
- Las unidas por e(14) (de la cadena principal)
- las que forman los puntos de ramificación, con es.(16)
- las de los extremos no reductores (OH del libre).
La 2,3,4,6 tetrametil-glucosa corresponderá a la del extremo no reductor, pues está unida a la cadena por el y tiene libres los C 2,3,4 y 6.
Por cada extremo no reductor tenemos una ramificación y podemos considerar que hay igual cantidad de puntos de ramificación que extremos no reductores. Estos puntos de ramificación tienen libres los C 2 y 3, y por tanto obtendremos 2,3 dimetil-glucosa.
Las glucosas de las cadenas tienen libres los C 2,3 y 6 y nos darán 2,3,6 trimetil-glucosa.
Nos queda la glucosa del extremo reductor, que nos dará 2,3,6 trimetil-glucosa, y no del 1, porque se elimina en la hidrólisis ácida.
Para calcular que % de cada uno haremos lo siguiente: como tenemos un total de 32,4gr de amilopectina,
como los extremos no reductores hemos visto que nos darían 2,3,4,6 tetrametil-glc y hemos obtenido 10,
200 100% x = 5% de extremos no reductores, y
10 x% consecuentemete lo mismo de puntos
de ramificación.
Partiendo de esto, y sabiendo que el número de glucosas que tenemos es , el 5% de estas es 3700 ramificaciones por molécula de amilopectina.
También hay otro 5 % de extremos.
El % de enlaces (14) es el otro 90% .
TEMA 10
LÍPIDOS : ÁCIDOS GRASOS
Con el término “lípido” nos referimos a una sustancia de origen natural, insoluble en agua, pero sí en solventes orgánicos apolares como cloroformo o éter, que se caracteriza por una gran diversidad tanto estructural como fucional.
En cuanto a las funciones, destacamos que :
-
Son componentes de membranas.
-
Actúan como formas de almacenamiento de C y energía.
-
Son precursores de otras sustancias importantes en biología.
-
Actúan como aislantes para choques mecánicos, térmicos o eléctricos.
-
Constituyen recubrimientos protectores que impiden infecciones y pérdidas o entradas excesivas de agua.
-
Algunos son hormonas o vitaminas.
Clasificación
Son un grupo compuesto por una gran cantidad de biomoléculas, y hay muchas clasificaciones. Seguiremos la siguiente:
- simples
lípidos - compuestos
- derivados
los lípidos simples están formados por ésteres de ácidos grasos y un alcohol únicamente. Dentro de estos, según cual sea el alcohol y los ácidos grasos, tendremos acilgliceroles o ceras.
Los lípidos compuestos además tienen ésteres formados por otro tipo de compuestos. Los subdividimos en fosfolípidos (con ác. fosfórico), y glucolípidos (cerebrósidos y gangliósidos) .
Los fosfolípidos se subdividen en fosfoacilgliceroles (su alcohol = glicerol) y en esfingolípidos.
Además, hay un tercer grupo, los derivados, que no se pueden incluir en los anteriores. En la composición de éstos no intervienen ácidos grasos, aunque algunos derivan in vivo de ellos.
- acilgliceroles
- Simples - ceras
- fosfolípidos (fosfoacilgliceroles y esfingolípidos)
- Compuestos
Lípidos - Derivados - glucolípidos (cerebrósidos y gangliósidos
lípidos simples: sus ácidos grasos son ácidos carboxílicos alifáticos de cadena larga (se llaman ácidos grasos por estar en las grasas). Como características generales de los más abundantes en la naturaleza podemos decir que:
-
la mayoría son ácidos monocarboxílicos (1 COOH)
-
las cadenas de hidrocarburos suelen ser lineales, no ramificadas y con un número par de átomos de C, que oscila entre 12 y 20
Con frecuencia aparecen dobles enlaces (llamados insaturaciones). La insaturación es frecuente a partir de los 16 átomos de C. Los dobles enlaces de los ácidos grasos suelen tener la configuración CIS, y pueden presentarse varios la misma molécula. Cuando esto ocurre, los dobles enlaces nunca están seguidos, sino separados por un solo grupo metileno.
La larga cadena hidrocarbonada es apolar, pero la molécula del ácido graso contiene un grupo polar, el COOH, y por tanto adquiere polaridad en conjunto. De echo, los ácidos grasos son ácidos débiles cuyos valores de pK están situados en 4,5 por término medio. Esto quiere decir que a pH fisiológico, se encuentran en forma aniónica (), con lo que es muy hidrófilo, mientras que las colas hidrocarbonatadas son hidrófobas.
Por tanto, los ácidos grasos se comportan como moléculas anfipáticas típicas, que, al intentar disolverlas en agua, tienden a formar monocapas en la interfase aire-agua con los grupos (carboxilato) sumergidos en el agua y las colas no polares fuera de ésta.
Si agitásemos esta mezcla, los ácidos grasos formrían las denominadas “micelas”, en las que las cadenas hidrocarbonadas se agrupan hacia el interior y los grupos carboxilato se dirigen hacia el exterior acuoso.
Si se mezclan con agua y con una sustancia oleosa, las micelas se van a formar sobre las gotas de aceite, emulsificándolo, y esta es la base del funcionamiento de los jabones, ya que estos son sales de ácidos grasos.
Nomenclatura
Habitualmente es por su nombre común (ácido esteárico, palmítico, linoleico...), pero además, existe un nombre abreviado que consiste en poner el número de átomos de C y a continuación, si hay dobles enlaces, indicamos la posición y número de estos. Por ejemplo,
Ácido palmítico: C16 : 0
Ácido esteárico: C18 : 0
Ácido oleico: C18 : 19 ó C18 : tiene un doble enlace en el
Ácido linoleico: C18 : 29,12 ó C18 :
La numeración de los átomos de C comienza por el grupo funcional, excepto un caso, la nomenclatura relacionada con el ámbito de la nutrición, que lo hace al revés. Se considera que el grupo metilo terminal tiene la posición 1 y se designa por la letra omega ()
Se suelen representar así:

En el laboratorio se separan e identifican por medio de la cromatografía de gas-líquido (lo más frecuente). Para poder separarlos, el requisito es que sean volátiles, y hay que transformarlos, pues, en derivados volátiles, haciéndolos reaccionar con metanol para que se formen los ésteres metílicos correspondientes que sí son volátiles.
Al principio, salen en el cromatograma los ácidos grasos (realmente sus ésteres) más pequeños, con menos átomos de C.
En cuanto a los dobles enlaces, salen antes aquellos que posean menor número de estos con igual número de carbonos.
Reacciones de los ácidos grasos
Una importante es la peroxidación (formación de peróxidos), que puede ser encimática o no enzimática.
En la no encimática, los agentes son oxidantes fuertes como el agua oxigenada () o el radical hidroxilo, que atacan a los dobles enlaces de los ácidos grasos, que son muy susceptibles a estas reacciones. Los compuestos que se forman son los hidroperóxidos.
Esta reacción es dañina para el organismo porque afecta a los lípidos de membrana, y es causante de enranciamiento de alimentos.
Aquí, se puede causar la oxidación de proteínas de membrana con el consiguiente efecto sobre la estructura o funcionamiento de la membrana.
Las células de los organismos vivos poseen mecanismos para protegerse de estas reacciones de peroxidación, y entre estos, tenemos la existencia de agentes como la vitamina C o la E que funcionan como anti-oxidantes, que hacen que ellas mismas sufran la reacción y así evitan que la sufran otras biomoléculas.
La peroxidación puede estar también catalizada encimáticamente. Esto ocurre, por ejemplo, en la formación de algunos lípidos derivados de ácidos grasos como la prostaglandina, que en su formación intervienen peroxidaciónes catalizadas por enzimas.
El envejecimiento celular está también relacionado con la formación de peróxidos de ácidos grasos.
Prostaglandinas
Su nombre procede de “próstata”, donde fueron aisladas por primera vez, pero se encuentran ubicadas en diferentes partes del organismo.
Estos lípidos presentan gran iportancia en medicina por sus múltiples efectos fisiológicos, entre los que destacan la contracción del músculo liso y la disminución de la presión sanguínea, que las ejercen a concentraciones muy bajas.
Se sintetizan in vivo a partir de ácidos grasos insaturados, y algunos medicamentos anti-inflamatorios como la aspirina inhiben su formación, ya que intervienen en el dolor.
En medicina se emplean en la inducción del parto e inhibición de secreción gástrica.
Estructuralmente todas derivan del Ácido 15-hidroxi -13- trans - prostenoico, un ácido graso de 20 átomos de C que tiene un doble enlace extra que provoca que se forme un anillo de ciclopentano que se forma entre los C 8 y 12, con un OH en el

Todas las prostaglandinas poseen este anillo, y las diferentes familias se diferencian entre sí según los sustituyentes oxigenados de ese anillo de ciclopentano.
Las familias son la A, B, E y F
La A un oxígeno unido al anillo con doble enlace
La B además, 1 doble enlace en el anillo
La C la A + 1 OH
La F la A + 2 OH
Dentro de cada familia se diferencian según el número y tipo de dobles enlaces de la cadena lineal, así como los sustituyentes que haya sobre esta cadena lineal.
TEMAS 11 Y 12
GLICÉRIDOS Y CERAS. FOSFOGLICÉRIDOS Y ESFINGOLÍPIDOS
Tanto los glicéridos (o acilgliceroles) como las ceras son , como vimos en la clasificación anterior, lípidos simples.
Los glicéridos son ésteres de la glicerina (o glicerol), un tri-hidroxi-alcohol + 1,2 ó 3 ácidos grasos. Así tenemos los mono, di o triacilgliceroles. Ejemplo:

glicerina monoacilglicerol
Los más abundantes en la naturaleza son los triglicéridos. Según los ácidos grasos que los formen, diferenciamos los simples de los mixtos (en los di o tri). En los simples, todos los ácidos grasos son iguales, y en los mixtos, distintos.
Todos los acilgliceroles son moléculas neutras (sin grupos polares). Además, según el estado físico a temperatura ambiente, diferenciamos las grasas y aceites (sólidos y líquidos respectivamente).
En general, el punto de fusión es menor cuanto mayor es el contenido en ácidos grasos insaturados.
El átomo de C central en el glicerol es quiral, asimétrico, y por tanto, estos lípidos presentan isomería óptica. La mayoría son isómeros “L”.
Los acilgliceroles se nombran de 2 maneras, usando el nombre del ácido graso + “-IL” e indicando la posición en que está unido el ácido graso (Con más de 1 ác. graso igual, y si son iguales, podemos decir, por ejemplo, di-oleil-glicerol, tri-palmitoil-glicerol).
La otra manera es con el sufijo “-INA”. Por ejemplo, con un oleico y dos palmíticos, diríamos momo-oleil,di-palmitoina.
Características
-
Son insolubles en agua, como todo lípido.
-
Constituyen los depósitos de grasa del tejido adiposo (triglicéridos), como almacenes de energía y carbono.
-
Se transportan a través de los vasos sanguíneos y linfáticos gracias a la formación de partículas lipoproteínicas como los quilomitrones.
-
Protegen física y químicamente a los órganos del cuerpo.
-
Todas las características generales de los lípidos.
Reacciones
La reacción más importante de este grupo es la hidrólisis. La hidrólisis no encimática en medio ácido, no va a dar lugar al glicerol + ácido graso. Si es en medio básico (presencia de sosa o potasa) obtenemos el glicerol + sales sódicas o potásicas de los ácidos grasos (jabones). Esta reacción, por tanto, se denomina “ de saponificación”.
En las células también tiene lugar la hidrólisis de los acilgliceroles y está catalizada por encimas (lipasas) que producen el glicerol y ácidos grasos. Estas muestran especificidad de enlaces (de los 3 enlaces éster que pueden existir en un triglicérido).
Las ceras también pertenecen al grupo de los lípidos simples, y son ésteres de un alcohol y de un ácido graso, pero su característica es que ambos son de cadena larga.

Como con los acilgliceroles y casi todos los lípidos, las ceras también son insolubles.
En el caso concreto de las ceras, estas sirven como capas protectoras de piel y plumas en animales, así como hojas y frutos en plantas. Protegen de infecciones y de pérdidas de agua.
Entramos ahora en el tema 12, y con él, en los lípidos compuestos. Como habíamos visto, los compuestos son:
Fosfoacilgliceroles fosfatos Esfingomielinas
Lípidos compuestos: Cerebrósidos con esfingosina
Gangliósidos
Los fosfoacilgliceroles (o fosfoglicéridos)
Son formadores de membrana. El más sencillo es el ácido fosfatídico, un glicerol unido enlaces éster a 2 ácidos grasos y 1 ácido fosfórico. Es precursor de todos los demás fosfoglicéridos.
Todos los fosfoacilgliceroles se van a diferenciar en que al ácido fosfórico se puede unir por un enlace éster a otro compuesto que va a distinguir los tipos.
Los ácidos grasos no nos permiten diferenciar tipos. Lo que sí podemos decir es que, en general, el ácido unido al suele ser saturado, y el del insaturado.
Este radical identificador puede ser la colina, una base nitrogenada con un grupo OH con el que se une al ácido fosfórico. Cuando es así, el fosfoacilglicérido resultante es la “fosfatidilcolina” o “lecitina”.
Si el radical es la etanolamina, el resultante es la fosfatidiletanolamina o cefalina.
Si es el aminoácido serina, se forma la fosfatidilserina.
Estos grupos son grupos cargados, y por tanto los fosfoacilgliceroles serán moléculas anfipáticas.
El radical R puede ser el glicerol, formándose el fosfoacilglicerol “fosfatidilglicerol”.
La R puede ser también un fosfatidilglicerol, y al unirse a otro ácido fosfatídico resulta un difosfatidilglicerol:
ác. fosfatídico + glicerol + ác. fosfatídico
también puede ser un alcohol cíclico, el más abundante es el inositol. En este caso se formaría el fosfatidilinositol, en el que a su vez el inositol puede llevar unidos también grupos fosfato.
Características
Estos lípidos se caracterizan porque son moléculas anfipáticas, ya que las largas cadenas hidrocarbonadas de los ácidos grasos unidas al glicerol forman la parte hidrofóbica de la molécula, mientras que el ácido fosfórico y la molécula a la que va unido (cargada) constituyen la cabeza polar hidrofílica de la molécula.
En el laboratorio podemos determinar cual es la estructura de un fosfoacilglicerol. Para ello usamos unas encimas que se llaman fosfolipasas que existen en las célula y son responsables del recambio de los fosfoacilgicéridos de las mismas y son específicas también para cada enlace. Cada una rompe distintos enlaces éster.
- La fosfolipasa A1 rompe enlaces de ácidos saturados
- La fosfolipasa A2 repme los de ácidos insaturados
- La D , los de ácidos fosfóricos con otras moléculas
En el laboratorio hidrolizamos con estas encimas, recogemos los resultados y determinamos la molécula de inicio.
Existen algunas variantes de los fosfoacilgliceroles, de entre los cuales vamos a destacar los que tengan 1 o las 2 cadenas hidrocarbonadas unidas al glicerol por enlaces éter, no éster.
Pues bien, cuando además se introducen insaturaciones, lo que obtenemos son los “plasmalógenos”, lípidos importantes que forman parte de algunas membranas plasmáticas como las de las células del corazón.
Los Esfingolípidos
Se caracterizan porque están formados por un alcohol que es la esfingosina, un alcohola aminado ( ) de 18 Carbonos.
El esfingolípido más simple, y precursor de los demás, es la ceramida. 1 ceramida = 1 ácido graso + esfingosina, solo que la unión no es por un enlace éster, sino por un enlace amida, establecido entre el grupo amino de la esfingosina y el ácido carboxílico del ácido graso.
Por unión de la fosfocolina (colina + P) lo que obtenemos es otro tipo de lípidos, las “esfingomielinas”. Aquí la fosfocolina se va a unir al OH terminal de la esfingosina. Las esfingomielinas también poseen naturaleza anfipática. Poseen una cabeza polar y cola no polar.
Estas sustancias las encontramos en las membranas celulares de los nervios y del encéfalo.
Por unión de la ceramida a un glúcido, entonces tenemos los glucolípidos, que también son anfipáticos, y que los encontramos en las membranas celulares del tejido nervioso y encéfalo. Los más sencillos son los cerebrósidos, ya que en ellos, es un monosacárido el que se une al OH terminal de la ceramida, que puede ser Glc o Gal. Formándose, respectivamete, glucocerebrósidos y galactocerebrósidos.
El monosacárido que forma los cerebrósidos puede estar sulfatado (). A este tipo se les denomina lípidos “sulfátidos”. Esto es más frecuente con la galactosa.
En el caso de los gangliósidos, los acúcares unidos son oligosacáridos, no monosacáridos, y además son unos que se caracterizan por llevar al menos un residuo de ácido siálico.
Además de dar rigidez a las membranas, los gluucolípidos presentan otras funciones a nivel de superficie celular, como el servir de marcadores antigénicos o marcadores para identificar el estado de diferenciación celular. También participan en el control del crecimiento de la células, y se relacionan con la transformación de células normales en cancerosas,y también en la unión de células con ciertas toxinas.
TEMA 13
TERPENOS Y ESTEROIDES
En este capítulo hablaremos de los lípidos derivados del ISOPRENO. El isopreno es el 2-metil-butiadieno. Éste polimeriza para dar lugar a una gran variedad de moléculas como los terpenos y esteroides, pero también otros lípidos isoprenoides. El mayor polímero del isopreno es el caucho, sacado de determinados árboles.
Los terpenos son hidrocarburos o alcoholes formados por varias unidades de isopreno, con modificaciones mínimas. El aroma característico de muchos frutos y plantas se debe a estos compuestos.
Los terpenos más simples son los monoterpenos, formados por 2 unidades de isopreno.
Los siguientes son los diterpenos, formados por 4 unidades, y entre los que destaca el fitol, alcohol de la clorofila (con 20 C).
Tenemos también triterpenos, con 6 unidades de isopreno y 30 carbonos, de entre los que destaca el “escualeno”, precursor del colesterol.
Otra familia de terpenos son los “dolicoles”, formados por también 20 unidades de isopreno, y que se encuentran en las membranas del R.E. y el Aparato de Golgi. Fosfatados en un extremo, sirven de transportadores de las proteínas durante su glicosilación. El dolicolfosfato es el encargado de este transporte.
Los esteroides son el grupo más grande, y se caracterizan por contener en su estructura un sistema de anillos que es el “ciclopentanoperhidrofenantreno”, del cual derivan.
Este sistema de anillos está formado por 17 C cuya numeración es específica. Además lleva una serie de sustituyentes de entre los que destacan 2 grupos metilo sobre los C 10 y 13, y numerados como 18 y 19. estos metilos están presentes en todos los esteroides excepto en los estrógenos, los cuales solo poseen uno.
Los esteroides llevan también una cadena hidrocarbonada sobre el que se numeran sus carbonos desde el 20 en adelante. La longitud de esta cadena es variable. Puede no existir como en las hormonas sexuales o tener hasta 9 C. También pueden existir sustituyentes oxigenados y dobles enlaces en posiciones variables según el esteroide del que se trate.
En su estructura existen muchos carbonos asimétricos, por tanto el número de enantiómeros posibles es muy elevado, aunque en la naturaleza no se encuentran todos ellos. Así, en todos los enantiómeros conocidos, los metilos 18 y 19 se sitúan hacia arriba del ciclopentanoperhidrofenantreno. El resto de los sustituyentes si están en posición CIS con los metilos (del mismo lado) se denominan sustituyentes. Si por el contrario están en posición TRANS, son sustituyentes .
Los esteroides se suelen clasificar según la complejidad de la cadena lateral unida al . Así diferenciamos 5 grupos principales.
1º.- Los esteroles :
Contienen siempre algún grupo alcohol. El más importante y conocido es el colesterol, que tiene un OH en posición . Se caracteriza por un enlace doble entre los carbonos 5 y 6, y por la cadena lateral sobre el que contiene 8 átomos de C. El colesterol actúa como precursor de otros esteroides y está relacionado con enfermedades como la arteriosclerosis.
2º.- Los derivados de la vitamina D :
Se forman por acción de la luz ultravioleta sobre los esteroles. Su característica estructural principal es la apertura del anillo B con la formación de un doble enlace entre los C 10 y 19. la cadena lateral conserva la cadena lateral del esterol del que deriva. Pueden existir derivados hidroxilados de esta estructura en varias posiciones. Estos compuestos regulan el metabolismo del calcio y el fosfato.
3º.- Los ácidos biliares:
Se caracterizan por, primeramente, la presencia de varios hidroxilos que tienen configuración , y por tanto en trans con los metilos. Estos OH se sitúan en las posiciones 3,7 0 12, según el ácido biliar. Por ejemplo, en el ácido cólico hay OH en las 3 posiciones, y por ello es el más soluble. En otros ,como el desoxicólico, no hay el del , o el litocólico, con solo el del , siendo el más insoluble, y es responsable de los cálculos biliares.
Esta abundancia de OH's les confiere características anfipáticas, esenciales para su función de emulsionar las grasas de la dieta, facilitando así la acción de encimas digestivas.
Otra característica de los ácidos biliares es su cadena lateral sobre el , que siempre finaliza con un ácido graso y consta de 5 átomos de C.
En el organismo humano existen muy pocos ácidos biliares libres, sino que su grupo ácido suele estar unido con el grupo amino de los aminoácidos glicina o taurina. Los derivados son glicocólico,tauricólico...
4º.- los corticosteroides:
Se forman en la corteza y regulan gran cantidad de procesos fisiológicos. Por ejemplo, el cortisol se caracteriza, como todos, por:
-
tener 1 grupo “ceto” en posición 3
-
1 doble enlace entre las posiciones 4 y 5.
-
La cadena lateral sobre el es corta y suele constar de solo 2 átomos de C y contiene oxígeno (=O).
-
Poseen algún otro grupo hidroxilo que puede estar en posiciones variables, siendo las más comunes en los carbonos 11 y 17.
5º.- Las hormonas sexuales:
Hay dos grupos, las masculinas o “andrógenos” y las femeninas o “estrógenos”. En ambos casos no existe cadena lateral en el . Además en el caso de los estrógenos, el anillo “A” es aromático, y no existe el grupo metilo sobre el .
Otra característica es la presencia de funciones oxigenadas en los y .
A parte de terpenos y esteroides existen otros componentes isoprenoides con interés en biología como los carotenos, que son pigmentos de frutas y hortalizas rojas. Son precursores del retinol o vitamina A.
También son isoprenoides los tocofenoles, entre los que destacamos el -tocofenol o vitaminia E.
Otros son las plastoquinonas de la cadena transportadora de electrones fotosintética, así como la “ubiquinona” o coenzima Q de la mitocondria.
La vitamina K también la ubicamos aquí, y participa en la coagulación sanguínea.
PROBLEMAS:
El número yodo de un cierto ácido graso insaturado con 2 dobles enlaces es de 180. si el peso atómico del yodo es 127, ¿cual podría ser el ácido traso?
El número o índice de I es:
Gramos de yodo que adicionan 100 gramos de grasa o ácido graso. El yodo se adiciona a los dobles enlaces, de tal manera que a cada mol de dobles enlaces se le adiciona 1 mol de .
Sabemos que 100 gramos de ácido adicionan 180 de , y como 1 mol adiciona 1 mol de ...
Si I = 127 = 254 100 180
1 mol 254 gr de x 2 x 254
x = Pm ácido graso = 282 ácido linoleico
2.- Indicar de entre las siguientes grasas, cual tendrá un mayor índice de saponificación:
-
palmitoesteaooleina
-
triestearina
-
estearoolioinoleina
-
trioleina
-
estearodioleina
El índice de saponificación es la potasa en miligramos necesaria para saponificar un gramo de grasa.
Como 1 mol de triglicérido reacciona con 3 moles de KOH, que son x gramos de KOH,
la de menor peso molecular será la de mayor índice, pues cuanto más pequeño sea el Pm, más gramos de KOH le tocará por gramo de grasa.
La primera es la más pequeña por su número de átomos de C. Como todas las demás tienen ácidos grasos de 18 átomos de C, habría que ordenarlas según sus insaturaciones.
TEMA 14
AMINOÁCIDOS
Los aminoácidos son moléculas que contienen al menos 1 grupo amino y 1 grupo ácido, que suele ser un COOH, aunque puede haber excepciones. La taurina tiene un ácido .
La primera clasificación de los aminoácidos se basa en la posición relativa de los grupos ácido y amino.
El carbono al que está unido el ácido graso se llama , y a los demás,...
Cuando el grupo amino está unido al este aminoácido se denomina - aminoácido. Si el amino está unido al , es un - aminoácido...
Esta clasificación nos permite establecer que en el caso de los aminoácidos proteicos, todos ellos pertenecen a la serie . Los aminoácidos proteicos los clasificamos según su cadena lateral.
Al átomo de C al que están unidos el amino y el ácido, le quedan otros dos posibles enlaces que los ocupan 1 átomo de H y una cadena lateral (el triángulo grupo amino-ácido-H es común para todos los aminoácidos proteicos).
La clasificación más habitual establece 4 grupos:
-
aá. Hidrófobos o de cadena apolar
-
aá. Polares con carga neutra
-
aá. Aniónicos o ácidos
-
aá. Catiónicos o básicos
Los aminoácidos hidrófobos se caracterizan por tener cadenas laterales hidrocarbonadas con algún otro elemento como S o H que mantiene la apolaridad. Aquí incluimos:
-
glicina
-
alanina
-
valina con cadenas laterales alifáticas
-
leucina
-
isoleucina
-
fenilalanina
-
triptófano con cadenas laterales aromáticas
- metionina (con un S en su cadena lateral)
La hidrofobia es mayor cuanto más voluminosa sea la cadena lateral. En el caso de la glicina o glicocola, el más sencillo, aunque se considera dentro de los hidrófobos, realmente lo es muy poco, ya que predomina el carácter iónico de los grupos amino y carboxilo.
Dentro de los aminoácidos de cadena apolar, también está la prolina, un aminoácido especial, ya que es cíclico, con 3 metilos, pero uno unido al grupo amino, formando un ciclo. Esta característica interviene en las propiedades de las proteínas en las que en su constitución interviene la prolina.
La prolina puede llamarse también “iminoácido”.
Los aminoácidos con carga neutra contienen en sus cadenas laterales, grupos hidrófilos que aumentan su solubilidad, como por ejemplo los grupos OH de la serina, treonina o tirosina.
También son aminoácidos con carga neutra la cisteína, con un grupo tilo (SH), así como la asparagina y glutamina, amidas correspondientes a los ácidos aspártico y glutámico.
Los aminoácidos aniónicos o ácidos tienen en su cadena lateral un grupo ácido y son solamente 2, el ácido aspártico y el ácido glutámico, diferenciados porque el segundo tiene 1 metileno más en su cadena lateral.
Los aminoácidos catiónicos o básicos son 3, la histidina, lisina y arginina. Su carácter básico se lo deben a grupos nitrogenados de su cadena lateral, que están cargados positivamente.
Los aminoácidos tienen, además de su nombre, un código que es sus 3 primeras letras o una única letra, códigos usados para trabajar con secuencias de polipéptidos y proteínas.
Además de los 20 aminoácidos proteicos existen otros en las proteínas que derivan de estos, ya que se les introducen modificaciones una vez que la proteína ya está formada. Estas modificaciones tienen como objetivo adaptar más la proteína a su función y al medio ambiente en el cual tienen que desempeñar su función. Las modificaciones más frecuentes son:
-
Metilaciones: se producen sobre el N de las cadenas laterales, sobre todo en el caso de la histidina y lisina, pudiendo hacerlo en 2 y 1 posiciones respectivamente. Estos aminoácidos metilados son frecuentes en las proteínas musculares.
-
Acetilación: también se produce en el grupo amino, y es frecuente en el caso de la lisina. Es importante la acetil-lisina en el caso de las histonas
-
Hidroxilación: esta modificación es más frecuente en la prolina, en las posiciones 3 y 4. estas abundan en el colágeno.
-
Fosforilación: Aquí se une un grupo fosfato mediante enlace éster a un grupo OH de la cadena lateral de aminoácidos, por lo tanto los aminoácidos que se forman son los que tienen OH en su cadena lateral como la serina, teronina y tirosina.
Los tres son muy importantes ya que son reversibles, y regulan la actividad de muchas encimas, así como la transcripción de genes...
-
Ciclación: la más común es la que se produce entre el grupo amino y el COOH de la cadena lateral del ácido glutámico. El resultante es el ácido piroglutámico, presente en el inicio de proteínas protectoras
-
Hipermodificaciones: destacan las hormonas tiroideas T3 y T4, que son dímeros iodados de la tirosina. También nos encontramos en la elastina de los tendones estructuras de cuatro residuos de lisina.
También hay aminoácidos no proteicos. Los hay los más comunes son:
Dentro de los- aminoácidos, tenemos muchos ejemplos ya que son intermediarios de los procesos de síntesis o degradación de aminoácidos proteicos, y por tanto se van a diferenciar de los proteicos, por ejemplo porque contienen 1 metileno más en la cadena lateral como en la homoserina respecto de la serina o la homocisteína respecto de la cisteína, otros que contienen un eslabón menos en la cadena lateral como en el caso de la ornitina respecto de la lisina.
Las primeras participan en el metabolismo de la cisteína y serina, mientras que la segunda participa en el ciclo de la urea.
Dentro de los - aminoácidos el más común es la - alanina, que es igual que la alanina, pero con el grupo amino situado sobre el C. Esta forma parte del ácido pantoténico, una viatamina.
Dentro de los - aminoácidos destacamos el - amino - butírico, abreviadamente “GABA”, que es un neurotransmisor.
Propiedades de los aminoácidos
Diferenciaremos: - isomería
- comportamiento anfótero
- absorbancia en el ultravioleta
- reacciones coloreadas de identificación
Con respecto a la isomería, en todos los aminoácidos proteicos excepto la glicina, el C es asimétrico. Esta característica provoca la existencia de al menos dos estereoisómeros para cada aminoácido, que van a ser D y L según tengan la cadena lateral a la derecha o a la izquierda.
La inmensa mayoría de los naturales son L - aminoácidos. Solamente nos encontramos trazas de aminoácidos D en las paredes celulares de bacterias en regiones muy determinadas, limitadas y minoritarias de las proteínas.
Que un aminoácido sea D o L va a condicionar el plegamiento de la proteína de la que forma parte.
Hay dos aminoácidos que poseen 2 centros quirales que son la treonina y la isoleucina, que tienen otro C también asimétrico, por tanto aquí, en estos aminoácidos va a haber 4 estereoisómeros posibles, 2 de la serie D y otros 2 de la serie L. Cuando existen dos estereoisómeros, el L nunca aparece, y se denomina “alo-”.
La absorbancia en ultravioleta aparece en la fenilalanina, tirosina y triptófano. a 280 nm se usan para determinar proteínas. El resto de los aminoácidos que no son aromáticos, absorben en el ultravioleta a 214 nm, y ya no son usados para determinar proteínas.
Reacciones coloreadas de identificación: los aminoácidos no son moléculas coloreadas, pero existen reactivos que dan lugar a compuestos coloreados cuando se unen a los aminácidos y que se usan para cuantificar estos mediante colorimetría.
De estos, el más usado es la nihidrina, que proporciona un color púrpura al reaccionar con todos los aminoácidos.
Se suelen separar los aminoácidos por otras técnicas, como por ejemplo cromatografía y después se identifican con la nihidrina.
El carácter anfótero de los aminoácidos es fundamental para entender su comportamiento en disolución, así como los sistemas de separación de estos.
Son electrolitos débiles. El grupo ácido y amino son ionizables. Los valores de pK oscilan entre 2 y 3 normalmente para el grupo ácido y entre 9 y 10 para el amino.
Cuando el pH es menor que el pK, los aminoácidos se encuentran totalmente protonados, y lo contrario sucede a pH inferior.
A un pH próximo a la neutralidad, el grupò ácido va a estar cargado (), y el amino estará también cargado () debido a sus pKs, y el aminoácido presenta carga neutra por compensación interna de las cargas positivas y negativas. A esta forma híbrida se le denomina ión híbrido o “zwitterion”.
Los aminoácidos que poseen un grupo ionizable en su cadena lateral, presentan un tercer pK, el . Aquí tenemos los aminoácidos básicos cisteína y tirosina.
El valor del depende del tipo de radical. Cuando es un ácido, este pK también es ácido (3 - 4). Cuando es una base, es básico (10 - 12).
Casos que nos encontramos según el pH del medio:
Cuando el pH es muy bajo, inferior al pK1, los aminoácidos están totalmente protonados ( COOH y).
Aumentando del pH, el aminoácido se disocia. El primer grupo que pierde el protón es el ácido, quedando como , y la molécula queda neutra ( y ). Como el pK1 oscila entre 2 y 3, a pH menor que pK1 el amino ácido estará en esta forma.
Aumentando más el pH se disocia el grupo amino, quedando la molécula con carga negativa ( y ). Esto se corresponde con el pK2, comprendido entre 9 y 10. A pH mayor a este, el aminoácido estará con esta forma.
Se define el punto isoeléctrico de un aminoácido como el pH en el cual la carga es neutra. Aquí se cumplen 2 condiciones: - la especie neutra es la predominante.
- las 2 formas restantes esán en equilibrio con la forma neutra.
El punto isoeléctrico puede calcularse para el caso de los aminoácidos neutros como la media del pK1 y el pK2.

Aminoácidos con un tercer pK:
En los equilibrios ácido - base de estos, al igual que antes, si partimos de pH < pK1, y con un aminoácido ácido, tendremos el aminoácido totalmente protonado, a medida que aumentamos el pH, se produce la disociación de los grupos en orden creciente de sus pK's: primero el ácido, después el ácido del radical, y por último el amino.
Con los aminoácidos básicos pasa casi lo mismo. Primero se disocia el grupo ácido, luego el amino, y luego el básico del radiacal, normalmente.
En los aminoácidos ácidos y básicos, el punto isoeléctrico se corresponde con la media de los 2 pK`s a ambos lados de la especie con carga neutra (pK1 y pKR en ácidos, y pK2 y pKR en básicos).
A pH próximo al punto isoeléctrico, la concentración del tercer grupo es muy baja, y no la tenemos en cuenta. Aquí, al igual que antes, en pH cercano al p.i., la forma neutra predomina, las de los lados están en equilibrio, y la otra se desprecia.
Todo esto es importante desde el punto de vista fisiológico ya que la actividad de muchas proteínas dependen del estado en el que se encuentren sus aminoácidos (disociados) así como las estructuras tridimensionales de las mismas.
TEMAS 15 a 21
PÉPTIDOS Y PROTEÍNAS
El enlace peptídico, que une aminoácidos para formar péptidos y proteínas, químicamente es un enlace de tipo amida, que se forma por condensación entre el grupo carboxilo y amino de otro con liberación de una molécula de . Lo que se forma es un dipéptido.
Con otro aminoácido más tendríamos un tripéptido, y así sucesivamente (tetrapéptido...).
Los péptidos se nombran empezando por el aminoácido con el grupo amino libre y acabando con el que tiene el grupo carboxilo libre (terminal). A los aminoácidos se les coloca el sufijo “-IL” excepto al último. Si son largos, se escribe la secuencia con las abreviaturas o bien con nombres propios.
El enlace peptídico tiene unas características muy importantes porque de ellas dependen las características del péptido y las proteínas.
-
En primer lugar, es un enlace muy resistente a la hidrólisis, con lo que las proteínas son muy estables.
-
Tanto el enlace C-O como el C-N tienen carácter parcial de doble enlace. Esto impide la libre rotación de la molécula alrededor del enlace peptídico, y como resultado, los 6 átomos que participan en el enlace son “coplanarios”, de tal manera que como las proteínas se pliegan para formar la estructura secundaria, lo que gira son los planos sucesivos de los enlaces peptídicos.
Partiendo de los 20 aminoácidos proteicos, el número de péptidos posibles es enorme, de echo, la variedad existente es muy grande.
Dentro de los péptidos de importancia biológica destacaremos en primer lugar el “glutatión”, un tripéptido que es el - glutamil - cisteinil - glicina.
Es muy importante porque actúa como protector de las condiciones reductoras en el citosol celular, ya que la cisteína posee un grupo SH y el glutatión puede formar dímeros por puentes disulfuro, pasando de la forma oxidada a la reducida.
Es uno de los péptidos más pequeños con interés fisiológico.
Las encefalinas son más grandes. Hay la Met-encefalina y Leu-encefalina, llamadas así porque se diferencian solo en el aminoácido carboxilo-terminal.
Son pentapéptidos (5) y se encuentran en el cerebro donde intervienen en procesos relacionados con la percepción del dolor.
También tenemos hormonas como la vasopresina y oxitocina, nonapéptidos (9). La primera regula la reabsorción renal de agua y la segunda la secreción de leche.
De mayor tamaño tenemos oligopéptidos de hasta 34 aminoácidos con función hormonal.
Un polímero de aminoácidos deja de ser un péptido para pasar a ser considerado proteína según dos criterios:
-
en primer lugar, la longitud (hasta 50 aá. Es un péptido) esto es bastante ambiguo, debido a que por ejemplo, la hormona paratiroidea, con 84 aminoácidos suele considerarse un péptido.
-
En segundo lugar, y más importante, la existencia biológica.
La cadena de aminoácidos que se sintetiza en el laboratorio y cuya existencia no ha sido demostrada en un organismo no será proteína por muy larga que sea. Si existe en la naturaleza, ello dependerá, ahora sí, de su longitud. Aún así, el límite es difuso.
Clasificación de las proteínas
Las proteínas se pueden clasificar según varios criterios, y todos son complementarios. Nos centraremos en 4 criterios:
-
Composición
-
Forma
-
Solubilidad
-
Función
- Según la composición, estas pueden ser simples o conjugadas. Las simples están constituidas solo por aminoácidos y las conjugadas contienen algún otro componente de diferente naturaleza química.
- Cuando este componente es un glúcido, son glicoproteínas.
- Si es un lípido, lipoproteínas.
- Si es un ácido nucleico, nucleoproteínas
otra variedad son los grupos prostéticos como el grupo “hemo”, que da hemoproteínas, o un metal, que da metaloproteínas...
- Según su forma, clasificaremos a las proteínas en globulares y fibrosas. Las primeras tienen forma esférica u ovoide, y suelen ser solubles, mientras que las segundas son alargadas e insolubles, constituyendo la base de los tejidos estructurales, por lo que también se las denomina escleroproteínas.
Como las globulares son solubles y las fibrosas no, podemos decir que la siguiente clasificación está relacionada con esta y puede considerarse como una subdivisión.
- Según la solubilidad, diferenciamos entre:
-
Albúminas, que son solubles en agua fría. Estas 2 tienen
-
Globlulinas, que son solubles en disoluciones salinas diluídas origen animal
-
Glutelinas, que son de origen vegetal, insolubles en agua y disoluciones salinas, pero sí en ácidos y bases diluidos.
-
Prolaminas, son de origen vegetal, y solubles en alcoholes de bajo peso molecular.
-
Protaminas, proteínas básicas que se encuentran en los fluidos seminales y son solubles en agua y

diluído.
-
Histonas, que interaccionan con el DNA en los núcleos y son básicas, solubles en agua y ácidos diluídos.
Dado que este grupo es de proteínas solubles, todas ellas serán, pues, globulares.
- Según su función (esta clasificación es la más heterogénea y la más informativa), pueden ser:
-
Encimas, que catalizan las reacciones biológicas
-
Transportadoras, como la hemoglobina (de

) o proteínas de membrana (de metabolitos)
-
De reserva, como almacén de energía, como la ovoalbúmina.
-
Contráctiles, como la actina y miosina, que intervienen en la contracción muscular
-
Estructurales, que son las más abundantes, de entre las que destaca el colágeno.
-
De defensa
-
Hormonal
-
Como factores tróficos. Se desempeña con el desarrollo de tejidos a nivel embrionario.
-
Receptores, de entre los que destacan los hormonales, proteínas de membrana a las cuales se une una hormona. Esta unión desencadena una acción hormonal.
-
Proteínas cromosómicas como las histonas
-
Toxinas, que abundan en microorganismos, insectos y reptiles
Estructura de las proteínas
Las proteínas son macromoléculas, en general bastante grandes (5000 aá. pequeña), con una estructura tridimensional muy compleja cuya determinación es una tarea difícil. Existen métodos como la cristalografía por rayos X, y en la actualidad se conoce menos del 10% de las estructuras de las proteínas aisladas de organismos.
Las posibilidades que tienen los aminoácidos de plegarse son grandes, pero cada proteína adopta una estructura espacial única, que se conoce como estructura nativa, y que está directamente relacionada con la función biológica de esta, de manera que si se altera esta estructura nativa, se ve alterada su función.
Según las relaciones entre estructura y función, tenemos 3 tipos:
-
proteínas homólogas
-
isólogas
-
análogas
Las homólogas, perteneciendo a diferentes especies, presentan similar función y similar estructura.
Las isólogas, van a ser originarias de la misma especie, desempeñan una función similar, pero tienen diferencias estructurales que las hacen más adecuadas a funciones específicas de esos organismos.
Las análogas también son originarias de la misma especie, su estructura es similar pero con diferencias en su función. En este caso, se cree que sus genes derivan de un gen ancestral común por duplicación y mutación.
El estudio estructural se realiza estableciendo 4 niveles que son las estructuras primaria, secundaria, terciaria y cuaternaria. Seguiremos estos cuatro niveles.
La estructura primaria:
Es la secuencia de aminoácidos. Nos indica el orden en el que están unidos los aminoácidos en la cadena polipeptídica.
En las proteínas conjugadas, nos indica también los lugares de unión con glúcidos o diferentes grupos prostéticos.
En la estructura primaria están las claves o información básica que determina las estructuras superiores, pero de momento no podemos predecir la estructura secundaria de las proteínas a partir de la primaria, nada más que e regiones pequeñas de estas proteíanas.
Esquemáticamente, la estructura primaria puede definirse como largas cadenas zigzagueantes de enlaces peptídicos y de carbonos![]()
alternantes, estando las cadenas laterales de los aminoácidos a los carbonos![]()
y denominándose residuos aminoacídicos.
Existen varias técnicas diferentes para determinar la estructura primaria de la proteínas, una clásica, que es el “método de secuenciación de Edman”, que consiste en:
-
Primero tenemos que disponer de la proteína purificada, y si está formada por diferentes cadenas polipeptídicas enrolladas, separarlas.
-
Luego estas las rompemos en péptidos de 20 aminoácidos.
-
Seguidamente separamos estos péptidos cortos y los hacemos reaccionar con el fenilisotiocianato, que se une al aminoácido con posición aminoterminal específicamente, y da lugar a que el enlace que une este aminoácido con el siguiente, sea más sensible a hidrólisis, con lo que luego se somete a una suave, y solo se romperá este enlace peptídico.
-
Obtenemos el derivado del aminoácido (aá. + FITC) y un péptido con un aminoácido menos, con el que podemos realizar un segundo ciclo de secuenciación.
Los aminoácidos que separamos los podemos determinar con técnicas como la cromatografía.
Estructura secundaria:
Nos define las disposiciones regionales y con elementos de simetría que nos podemos encontrar en determinadas regiones en las cadenas polipeptídicas.
Las posibilidades son enormes, sin embargo existe una restricción muy importante a todas estas posibilidades, que consiste en la planaridad del enlace peptídico, de tal manera que los parámetros que cambian en la cadena son los ángulos de giro entre planos consecutivos de este péptido. Los valores de estos ángulos nos definen las estructuras secundarias.
Existen muchas estructuras secundarias, pero entre las más estables se encuentran la hélice , la lámina (o hoja plegada), y son las más estudiadas.
Hélice :
En esta disposición la cadena polipeptídica se enrolla sobre sí misma como la rosca de un sacacorchos. La rotación es hacia la derecha, y las cadenas laterales de lo aminoácidos se disponen hacia el exterior por el volumen que ocupan.
Cada aminoácido está girado 100º con respecto al anterior, lo que quiere decir que hay 3,6 residuos de aminoácido cada vuelta completa.
Cada cuatro aminoácidos los enlaces peptídicos se encuentran cercanos, pero en niveles diferentes de la hélice, por lo tanto se pueden establecer puentes de hidrógeno entre estos enlaces ya que los grupos C-O y N-H tienen posición trans.
Estos puentes de hidrógeno son la principal fuerza que estabiliza la estructura en hélice y son determinantes de lo que se denomina la longitud del paso de rosca, que es de 54 nm en la hélice.
En la hélice los puentes de hidrógeno se establecen entre el grupo C-O del aminoácido que ocupa la posición “n” y el grupo ácido del aminoácido con posición n+4.
Existen variantes de esta estructura. Destacaremos 2 comunes estudiadas en las proteínas, la hélice y la hélice .
En la hélice tenemos 3 residuos por vuelta ya que los puentes de hidrógeno se establecen entre el grupo C-O del aminoácido “n” y el N-H del aminoácido “n+3”.
En la hélice , más ancha, nos encontramos con 4,4 residuos por vuelta; aquí los puentes de hidrógeno se establecen entre el grupo C-O del aminoácido “n” y el N-H del “n+5”.
Lámina o lámina plegada:
Aquí la cadenas polipeptídicas se disponen manteniendo su estructura en zig-zag extendidas y en dos posibles disposiciones, la paralela, en la que las diferentes cadenas tienen el extremo amino y carboxilo del mismo lado, y antiparalela, con el grupo amino y carboxilo contrapuestos. Esta última, la antiparalela es más frecuente.
La lámina tiene una disposición más compacta que la hélice. En la lámina la estabilidad depende de puentes de hidrógeno. Aquí los puentes se establecen entre el O de un grupo C-O y el H de un N-H de la cadena paralela próxima, son puentes intercatenarios, mientras que en la hélice eran intracatenarios.
Los residuos se sitúan alternativamente hacia arriba o abajo dependiendo del plano definido por la lámina.
Los aminoácidos con cadenas laterales pequeñas son más compatibles con esta disposición, debido a que tienen menos impedimentos estéricos.
En el caso de las proteínas globulares puede suceder que las dos cadenas que interaccionan por puentes de hidrógeno pueden ser fragmentos de la misma cadena, por medio de un giro.
Estructura terciaria:
Define el conjunto de todas las interacciones tales como puentes de hidrógeno, enlaces disulfuro, enlaces de Van der Waals... que rigen el plegamiento de la cadena completa en el espacio, y engloba todas las estructuras secundarias que pueden existir en distintas zonas de la molécula.
Además, define otras zonas como las de “ovillo estadístico”, zonas de giro o torsión, donde las cadenas se pliegan sobre sí mismas
Destacaremos los giros , que son bastante bruscos, por el establecimiento de puentes de hidrógeno entre los grupos C-O del aminoácido “n” y el grupo N-H del aminoácido “n+3”, empleados para conectar láminas antiparalelas.
También existen estructuras al azar con plegamientos al azar.
En algunos casos la cadena polipeptídica puede tener varias zonas de plegamiento con elasticidad propia. A estas se les denomina “dominios”.
En proteínas fibrosas las estructuras secundaria y terciaria coinciden, las cadenas laterales se orientan todas igual. Un ejemplo es la -queratina del pelo, que sirvió para determinar las dimensiones moleculares de las hélices
Estructura cuaternaria:
Define las interacciones entre varias cadenas polipeptídicas para formar una proteína. Las proteínas que solo son una cadena, son monómeros, si tienen dos cadenas son dímeros, si tres, trímeros, tetrámeros....
Son homodímeros si las dos subunidades son iguales, y heterodímeros si son distintas. Las fuerzas que estabilizan la estructura cuaternaria son variadas y son las mismas que las que estabilizan la estructura terciaria (enlaces disulfuro, puentes de hidrógeno, interacciones iónicas...).
Es frecuente que en las proteínas oligoméricas puedan existir varias estructuras cuaternarias, varios estados conformacionales que afecten a la actividad de la proteína, como en las encimas alostéricas o la hemoglobina.
EL COLÁGENO
Es la proteína fibrosa más abundante del cuerpo y posee una estructura secundaria característica que es la hélice P.
El colágeno es la proteína base del tejido conjuntivo y el constituyente orgánico básico de los tejidos calcificados.
La estructura primaria es única, se caracteriza por tener un 30% de glicinas, y alrededor de un 20% de prolina e hidroxiprolina. Estas dos son características del colágeno. Solo la elastina, que es otra proteína de tendones y cartílago también contiene estos dos amionoácidos en esta proporción.
La estructura secundaria. La hélice P es más rígida y extendida que la hélice (con cada aminoácido ascendía 18 nm) y aquí 0,29 nm.
Esto se debe a:
-
la presencia de prolina e hidroxiprolina, aminoácidos cíclicos con un anillo del cual forma parte el N , que da lugar a que el anillo sea rígido estéricamente a la cadena polipeptídica,
-
a que al estar el N formando parte del anillo no puede formar puentes de hidrógeno por no tener hidrógenos,
-
y a que los OH's de la hidroxiprolina establecen estos puentes de hidrógeno que estabilizan a esta hélice P.
La hélice P es levógira, se enrolla formando una triple hélice dextrógira que forma el tropocolágeno, que presenta 2 nm de diámetro y 260 nm de longitud.
Uno de cada 3 aminoácidos son glicinas.
En el tropocolágeno uno de cada tres aminoácidos orienta su radical hacia adentro de la hélice, y solo la glicina puede hacer esto.
Otra característica del colágeno es que estas cadenas polipeptídicas pueden unirse por enlaces transversales con otras cadenas que se establecen entre los residuos de la lisina y el lisinal (derivado de la lisina).
Estos enlaces también están entre diferentes unidades de tropocolágeno que se asocian para formar las protofibrillas y fibrillas del colágeno. Esta unión también se establece entre enlaces de residuos de lisina y lisinal.
El colágeno es una glucoproteína, y existen diferentes tipos de colágeno que se diferencian entre sí por la composición de las cadenas polipeptídicas que los forman. Existen dos tipos de cadena, la y la , que se diferencian en la secuencia de aminoácidos, en las glicosilaciones y en el número de OH's de la prolina.
Estas características de los distintos tipos de colágenos adaptan a los mismos para su función en el cuerpo.
Comportamiento de las proteínas en disolución
La mayor parte de las proteínas se encuentran disueltas en los líquidos biológicos, donde cumplen su función. Estos líquidos son disoluciones salinas acuosas.
Las propiedades derivan de dos características de las proteínas:
-
Su naturaleza poliiónica
-
Su naturaleza macromolecular
Con relación a su naturaleza poliiónica, tanto lo extremos de la cadena polipeptídica como los residuos de los aminoácidos ácidos y básicos contribuyen a que la proteína tenga cargas en su periferia, que se comporte como un poliión. Por este motivo hay contraiones orgánicos en la disolución que rodean la proteína por atracción electrostática y dejan su carga neutra.
Los aminoácidos poseen sus constantes de ionización, pero al estar unidos a la proteína, la presencia de otros grupos cercanos hace que se modifique, de manera que el pK de los residuos varíe en unidad.
Las proteínas, al igual también poseen un punto isoeléctrico (p.i.) en el que para ese pH su carga neta es cero.
-
Si pH < p.i. la proteína tiene carga positiva
-
Si pH > p.i. la proteína tiene carga negativa.
El punto isoeléctrico depende de la composición de aminoácidos de modo que si el contenido en aminoácidos básicos y ácidos es parecido, el p.i. se encuentra entre 6 y 8, si predominan los aminoácidos ácidos el p.i. será más bajo, como en el caso de la pepsina, cuyo p.i. es 1,5. gracias a esto, la pepsina puede resistir y desenvolverse por el ácido jugo gástrico e hidrolizar proteínas de tipo neutro
Las histonas son básicas, y pueden unirse al DNA, cargado negativamente.
El carácter poliiónico hace que la solubilidad dependa del pH, contenido salino del medio...
Como norma general, una proteína es más soluble si las interacciones entre moléculas proteicas se reducen al mínimo.
Respecto al PH, la solubilidad es mínima en el p.i. ya que no existe repulsión entre las moléculas y no se solubilizan
Con respecto al contenido salino, el efecto es bifásico, a una cierta concentración de sales, estas actúan como contraiónes, mientras que a una concentración elevada insolubilizan las proteínas ya que los iones se rodean de una esfera de solbatación con moléculas de agua, y estas no están libres en la disolución.
Con respecto a la característica de ser macromoléculas, al serlo forman disoluciones coloidales, fundamentales para membranas semipermeables. No las pueden atravesar por su tamaño. Esto se usa también para diálisis.
Hacen también de receptores iónicos que atraen a pequeños iones.
Desnaturalización de proteínas
La desnaturalización es el proceso por el cual una proteína cambia su estructura nativa, lo que conlleva a la pérdida de su actividad biológica.
Es provocada por el calor, PH's extremos o por agentes que rompen enlaces no covalentes como los detergentes o la urea.
Las estructuras afectadas son la terciaria y la cuaternaria. La proteína se despliega y los residuos hidrófobos que están dirigidos hacia el interior de la proteína, al desnaturalizarse afloran al exterior con la consiguiente isolubilización de la proteína.
Solo en los casos más extremos se ve afectada la estructura secundaria, pero nunca la primaria. Para romper los enlaces peptídicos hay que recurrir a tratamientos más fuertes o digestión con encimas específicas (proteasas) como la pepsina.
Plegamiento de las proteínas ( Cómo adoptan su estructura en 3D)
El mecanismo es muy complejo, en general está dirigido por la tendencia a alcanzar estados de mínima energía.
Una fuerza muy importante es la hidrofobia de los residuos de los aminoácidos, que dirige el plegamiento, de forma que los residuos polares se colocan hacia la periferia de la proteína en contacto con la solución acuosa, mientras que los residuos apolares o hidrófobos tienden a protegerse del agua colocándose en el interior de la proteína.
El plegamiento no es espontáneo a partir de cualquier conformación una vez que la proteína está formada, lo que explica que gran parte de las desnaturalizaciones sean irreversibles.
Cuando la proteína está siendo sintetizada, existen otras proteínas que se llaman “chaperoninas”, que colaboran en que se pliegue correctamente la cadena en crecimiento. Para ello, se unen a la cadena desplegada impidiendo interacciones no deseadas, en un proceso ligado a la hidrólisis del ATP.
En el plegamiento intervienen otras proteínas encimáticas como “isomerasas de puentes de sulfuro”, etc.
TEMA 20 Y TEMA 21
PROTEÍNAS CONJUGADAS
Estas proteínas llevan asociados otros componentes de naturaleza no proteica que se clasifican según este componente.
Glucoproteínas:
- Colágeno ( ya estudiado).
- Inmunoglobulinas: Son anticuerpos que reconocen y se unen a los determinantes antigénicos, regiones del anticuerpo que provocan la respuesta inmune.
Dentro de las inmunoglobulinas existen 5 tipos: Ig A, D, E, G, M.
La inmunoglobulina G son las más abundantes al defenderse de un organismo extraño. Están formadas por cuatro cadenas polipeptídicas iguales 2 a 2. son 2 cadenas ligeras y 2 cadenas pesadas. Las uniones entre las distintas cadenas se establecen por puentes disulfuro, lo que hace que sean bastante resistentes a la disociación.
Las cadenas ligeras constan de dos dominios, uno variable y otro constante. Las pesadas constan de cuatro dominios, uno variable y 3 constantes. Estos dominios son zonas globulares que también se estabilizan por puentes disulfuro.
Los dominios situados en el extremo aminoterminal tanto de las cadenas ligeras como de las pesadas (el primer dominio) constituyen el centro activo de la inmunoglobulina. Son los encargados de reconocer al antígeno y unirse a él. Esto es posible porque presentan una gran variabilidad estructural. El resto de la molécula es constante, con escasa variabilidad.
En la región constante de las cadenas pesadas (el segundo dominio) van oligosacáridos unidos.
Las inmunoglobulinas poseen una estructura característica en “Y”. A esta región se la denomina región “bisagra” porque permite que la molécula se abra o cierre, facilitando así la unión con el antígeno.
Diferentes tipos de inmunoglobulinas:
En lo esencial se repite la estructura mencionada. Una diferencia es la composición de la cadena pesada, característica para cada una de ellas, las de la Ig A son , las de las D,...
Las cadenas ligeras son iguales para las 5 clases de inmunoglobulinas y pueden ser de 2 tipos, o (landa o kapa). Nunca tendremos una y una en la misma cadena pesada o las dos en las ligeras.
En las inmunoglobulinas A esta estructura básica se repite entre 1 y 3 veces. En el caso de otras se repite 5 veces.
Existen también diferencias funcionales entre las diferentes Ig's durante el proceso inmune.
Lipoproteínas:
Son las responsables del transporte de lípidos por la sangre. Presentan una estructura esférica cuya parte interna es de naturaleza oleosa, hidrófoba. Están formadas por lípidos apolares como los ésteres de colesterol o triglicéridos.
Están cubiertas por una monocapa de lípidos anfipáticos (fosfolípidos y colesterol libre), junto con las proteínas específicas que también son anfipáticas y dirigen sus residuos hidrofílicos hacia el exterior de la proteína y los hidrofóbicos hacia el núcleo hidrófobo de la proteína. A estas proteínas se las denomina “apoproteínas”.
La unión de los lípidos del núcleo a la capa externa de fosfolípidos y proteína es una unión débil no covalente, mediante puentes de hidrógeno y enlaces de Van der Waals, con lo que se permiten el intercambio de lípidos y apoproteínas entre las distintas lipoproteínas del suero y entre estas y los tejidos.
Se han identificado en el plasma sanguíneo 5 clases de lipoproteínas que se clasifican según su densidad y comportamiento en la electroforesis:
-
Quilomicrones: lipoproteínas muy ricas en triglicéridos procedentes de la dieta
-
Lipoproteínas de muy baja densidad: conocidas también como VLDL o lipoproteínas pre, según su comportamiento en electroforesis . también son ricas en triglicéridos, pero en este caso de síntesis endógena
-
Lipoproteínas de densidad intermedia
-
Lipoproteínas de baja densidad o LDL: estas transportan fundamentalmente colesterol. También se denominan lipoproteínas
-
Lipoproteínas de alta densidad o o HDL: los lípidos aquí son más abundantes, son fosfolípidos y colesterol.
La distinta densidad de las proteínas se debe a la distinta proporción de lípidos y proteínas de que consta. A mayor proporción de lípidos mayor densidad.
En cuanto a las apoproteínas, estas varían de unas a otras y se denominan como apoA, apoB, C, según el orden en que han sido descubiertas.
Que las apoproteínas sean tan distintas influye en el punto isoeléctrico de las mismas, y por tanto en su comportamiento en la electroforesis.
Hemoproteínas
Son aquellas que contienen el grupo hemo . como ejemplo, vamos a estudiar la mioglobina y la hemoglobina, que son proteínas que se unen al oxígeno.
Las hemoglobina transporta oxígeno en el interior de los glóbulos rojos de humanos y otros vertebrados, mientras que la mioglobina lo almacena en los tejidos hasta que es usado en el metabolismo.
La mioglobina se encuentra en concentraciones relativamente altas en el músculo esquelético y cardíaco, siendo la principal responsable de su color.
Representa el 8% de las proteínas musculares de los animales que se sumergen, que necesitan almacenar grandes cantidades de oxígeno para usarlo cuando están sumergidos.
La mioglobina es una proteína monomérica, mientras que la hemoglobina es un tetrámero.
En la mioglobina, la cadena polipeptídica está formada por 8 hélices en las cuales están más de las ¾ partes de los aminoácidos. Estas hélices se nombran de la A a la H comenzando por el extremo aminoterminal, y los segmentos interhélice se nombran AB,CD,DE...
En cuanto a los residuos, estos se pueden nombrar de 2 modos, desde el grupo aminoterminal al carboxiloterminal o bien A1, A2, A3... según la hélice donde se encuentren.
El oxígeno se une al grupo prostético de la mioglobina, que es el grupo hemo, una profirina , la protoporfirina 9. el oxígeno se va a unir a un átomo de hierro que se situa en el centro del grupo hemo, que es un sistema tetrapirrólico, formado por la unión de cuatro anillos derivados del tirrol, y que se caracteriza por los sustituyentes sobre este sistema de anillos, 2 vinilo, 2 propiónico (ionizables) y 2 metilo.
El grupo hemo se sitúa en una cavidad que forma la cadena polipeptídica de la mioglobina, de forma que los sustituyentes apolares se orientan hacia la región hidrófoba (interior de la proteína), mientras que los ionizables se dirigen al exterior acuoso. El hierro está unido en el centro del grupo hemo, por lo que los enlaces de coordinación.
4 de ellos se establecen con los cuatro nitrógenos de los anillos pirrólicos. El quinto enlace de coordinación lo establece con el N de un residuo de histidina denominado “Histidina Proximal”, con posición F8 o G3 de la cadena.
Un sexto enlace de coordinación se une al O. En la unión interviene la “histidina distal” (más lejana) A2 o E7.
El grupo hemo está oculto en el interior. Esto es esencial para su función, ya que el es capaz de unirse al O, mientras que si el grupo hemo estuviese en la solución acuosa tendría el hierro como , con lo que no podría unirse al O. Esta cavidad protege al Fe de una rápida oxidación.
Esta protección también tiene su importancia para la diferente afinidad que presenta para unirse con el CO, que compite con el O para unirse al grupo hemo, que cuando está libre, su afinidad por el CO es de 25.000 más que por el oxígeno, mientras que unido a la mioglobina, la afinidad por el CO es de solo 250 veces la del O.
La hemoglobina es un tetrámero formado por 4 subunidades , dos a dos iguales. Cada subunidad es igual a una mioglobina.
Existen diferentes tipos de hemoglobinas, según cuales sean los substituyentes que la formen. Las más abundante es la A1, formada por 2 cadenas y 2 . El 12% es del tipo A2, con 2 cadenas y dos cadenas. En el feto la más abundantes la HF, formada por 2 y 2 .
Unión del Oxígeno a la hemoglobina:
Cada molécula de hemoglobina puede unirse a 4 moléculas de O. Es una proteína alostérica. La unión es cooperativa ya que la unión del O a una subunidad favorece la unión a la siguiente y así sucesivamente. Esto es debido a que la unión proboca un cambio conformacional que se transmite a las otras unidades de la proteína.
Además, esta unión es reversible, depende de la concentración de O que existe en el entorno de la hemoglobina, temperatura, pH...
La importancia de la estructura 3D en su función:
Tomaremos como ejemplo las hemoglobinas anómalas. Estas son anormales, y provocan patologías. Una muy bien caracterizada es la hemoglobina S o falciforme, y los individuos que la poseen sufren un tipo de anemia “falciforme”.
Hay una mutación en los genes que codifican las cadenas que provoca que el aminoácido 6 de la cadena sea valina en vez de glutámico. A pH fisiológico el segundo está cargado negativamente y la valina no. Hay entonces una carga positiva que induce a que la hemoglobina precipite en el interior de los eritrocitos, y baja la concentración de hemoglobina en estado desoxigenado. Los glóbulos rojos que padecen esto tienen forma de hoz, y esto es una hemoglobinopatía.
TEMAS 22 a 25
NUCLEÓTIDOS Y ÁCIDOS NUCLEICOS
Los nucleótidos son un grupo de macromoléculas que participan en los procesos de transmisión y expresión de la información genética. Existen 2 tipos de ácidos nucleicos, el ác. ribonucleico (RNA o ARN) y el ác. desoxirribonucleico (DNA o ADN).
El nombre de los ácidos nucleicos hace referencia a su característica de ácidos, pues tienen ácido fosfórico, y la primera vez que se localizaron, fue en el núcleo de células eucariotas, pero sabemos hoy que también pueden existir en las mitocondrias, cloroplastos, y citoplasma, y también en todo procariota y virus.
Desde el punto de vista químico, los ácidos nucleicos son polinucleótidos, polímeros de nucleótidos que están unidos por un enlace característico, el fosfo-di-ester, que es un ácido fosfórico que se une por dos enlaces éster sucesivamente a dos grupos alcoholes.
Los nucleótidos están formados por tres componentes, que son el ácido orotfosfórico, una pentosa y una base nitrogenada.

Pentosas:
En el caso de los nucloeótidos de ARN (ribonucleótidos), la pentosa es la ribosa (ver temas de azúcares). En el ADN (desoxirribonucleótidos), las pentosas son la 2-desoxirribosa.
Ahora hablaremos de carbonos “prima” (1', 2', 3'...) para diferenciarlos de los C's de las bases nitogenadas (1, 2, 3...).
Bases nitrogenadas:
Hay dos tipos, las púricas y las pirimidínicas, según de qué anillo deriven, la purina o la pirimidina.
Los sustituyentes en el anillo determinan las bases.
-
Púricas: adenina y guanina
-
Pirimidínicas: timina (en ADN solamente),uracilo (solo en ARN) y citosina (en ambos).
Además de estas bases nitrogenadas, que son mayoritarias, existen otras minoritarias que algunas de ellas forman parte de solo unos tipos determinados de ácidos nucleicos, y otras que son productos intermediarios en el metabolismo de éstos.
Existen análogos sintéticos usados en terapia anticancerosa y antivírica.
Las bases nitrogenadas pueden existir en 2 formas que se llaman tautoméricas (ceto y enol). La primera predomina a pH fisiológico. En las “enol”, el doble enlace del oxígeno se transforma en sencillo. Que la base esté en una u otra forma influye en su capacidad para formar puentes de hidrógeno. Esto es de gran relevancia a la hora de estudiar ls estructura y función de los ácidos nucleicos, ya que estas bases nitrogenadas se emparejan entre sí por puentes de hidrógeno. En el caso del ARN esta molécula es monocatenaria pero pueden existir regiones bicatenarias por apareamientos de pares de bases complementarias.
Las bases se unen siempre una púrica con una pirimidínica, unión fundamental porque las primeras son grandes, con dos anillos, y las segundas pequeñas, con un anillo, lo que nos garantiza que el par de bases tiene el tamaño constante.
Se unen siempre G + C por 3 puentes de hidrógeno, una unión un poco más fuerte que la A + T (en ADN) o A + U (en ARN), formada por 2 puentes de hidrógeno.
En estos puentes de hidrógeno participan los grupos ceto. Si las bases están en forma enol, esto influye, y puede dar lugar a la formación de puentes de hidrógeno incorrectos.
Unión entre los constituyentes:
La pentosa y la base nitrogenada se unen por un enlace N-glucosídico, que se establece entre el C de la pentosa (el anomérico), con el N de las pirimidinas, o bien con el de las purinas. El compuesto resultante es un “nucleósido”. Entorno a este enlace hay posibilidad de giro, y esto hace que en los ácidos nucleicos puedan existir diferentes conformaciones según la posiciónes relativas que ocupan la pentosa y la base nitrogenada.
De entre estas conformaciones están la SIN y la ANTI, que dependen de cómo esté colocada la base nitrogenada con respecto a la pentosa (base hacia fuera de la pentosa o hacia adentro).
Si a un nucleósido le añadimos un ácido fosfórico mediante un enlace éster, tenemos un nucleótido. El ác. se une por enlace éster a las posiciones 5' o 3' (a los OH's). Aquí, al unirse una única molécula de ácido fosfórico, el resultante es un nucleósido monofosfato.
Denominación: (según la base y la pentosa)
Si el azúcar es la ribosa, y la base nitrogenada la adenina, el nucleósido resultante es la adenosina. El símbolo es la inicial de la base nitrogenada. Si agregamos un ácido fosfórico, obtenemos un nucleótido, ácido adenílico, uridílico, guanílico... . Se representan como los anteriores con la misma inicial y con una “p” minúscula a la izquierda, lo que quiere decir que el ácido fosfórico está unido al C 5' (también se pone 5'-AMP, y normalmete se suele decir AMP cuando es el 5'). Si el ácidos fosfórico se une en la posición 3', para diferenciarlo, colocamos la “p” en el lado derecho (o 3'-AMP).
Si la pentosa es una desoxipentosa, se añade “desoxi-“ a la palabra (desoxiadenosina, ácido desoxiadenílico...).
Hemos mencionado los nucleótidos monofostato. Existen también nucleótidos di y trifosfato, y momofosfato cíclicos.
Si al grupo fosfato en posición 5' le añadimos otro ácido fosfórico, tenemos un nucleótido difosfato, que lo indicaremos poniendo en la nomenclatura dos “p's” minúsculas o una D (ADP). Si añadimos otro ácido fosfórico a la molécula obtenemos un nucleótido trifosfato, como el ATP.
Los enlaces entre fosfato y fosfato son “anhídridos”.
Entre los monofosfato cíclicos está el AMP cíclico, en el que la molécula de fosfato se une a los carbonos 5' y 3' por dos enlaces éster.
Los nucleótidos mono, di, trifosfato y lo monofosfato cíclicos son importantes porque, además de formar los ácidos nulceicos, intervienen de forma libre en reacciones metabólicas y reacciones de intercambio de energía en la célula.
Los nucleótidos se pueden representar así:

Los nucleótidos se unen por enlaces fosfodiester para formar los polinucleótidos, y se establecen por un enlace éster en el grupo OH 3' de la ribosa, a al OH 5' de otra pentosa. En este momento tendríamos un dinucleótido. Aumentando el número de nucleótidos tendríamos trinucleótidos, tetranucleótidos...oligonulceótidos, polinucleótidos, ácidos nucleicos.
En los polinucleótidos tenemos un esqueleto de uniones azúcar-fosfato constante para el ADN y ARN (fosfato - pentosa, fosfato - pentosa....) siempre en el mismo sentido (5' 3'). Lo característico de la secuencia es como van situadas las bases de estos nucleótidos (G, A, T, U, C......) desde el nucleótido 5' fosforilado, hasta el 3' libre (con OH). Un ejemplo sería: CGUA. Si solo pusiésemos CGUA sobreentenderíamos lo demás.
Por apareamiento con puentes de hidrógeno se unen 2 bases complementarias, formando cadenas dobles como el DNA. Las cadenas de DNA son un poco más estables que las de ARN, sobre todo, respecto a la hidrólisis alcalina. Esto se debe a que en el DNA la pentosa es la desoxirribosa, y en esta no pueden formarse anillos por no tener OH sino H .
Que el ADN contenga timina en vez de uracilo también afecta a la estabilidad de este ácido nucleico, porque pueden producirse desaminaciones. Por ejemplo, la desaminación de citosina da uracilo.
Otra diferencia existente entre el ADN y al ARN es que en la composición de los ARN existen unas relaciones de bases nitrogenadas que no encontramos en los ADN's. En ADN, la concentración de Timina es igual a la de Adenina, y la [G] = [C].
El contenido de G y C varía de una especie a otra pero no en individuos de la misa especie. En el ARN la composición es más variable.
Estructura secundaria (3D) de los polinucleótidos
DNA:
La mayor parte de las cadenas de ADN son bicatenarias, con excepción de algunos virus. Estas dos cadenas discurren antiparalelas:

Dentro de ellas se encuentran los pares de bases unidos por puentes de hidrógeno de manera que siempre se juntan una púrica con una pirimidínica. El ancho es de 11 Å.
La forma mayoritaria es la de DNA-B o de “wattson y Crick” (1953 premio nobel). En esta, la estructura bicatenaria se enrolla en una doble hélice dextrógira , en la que se puede observar que cada vuelta de hélice se corresponde a 10 pares de bases. Hay una distancia de 3,4 Å entre cada par de bases, con lo que una vuelta de hélice tiene 34 Å. El ancho de la doble hélice tiene, también constante, 2nm.
Hay dos tipos de surcos, los “surco mayor” y “surco menor”. Aquí, los pares de bases están en el interior y los fosfatos en el exterior (cargados negativamente).
Estas moléculas, en disolución, presentan estructuras largas, flexibles, de aspecto filamentoso, que se vuelve más compacto cuando la molécula reacciona con otros componentes de la célula, como proteínas.
Además de esta forma de ADN-B existen otras formas isoméricas. Las más estudidadas son el ADN A y el Z.
Del ADN-A todavía no se conoce bien su función. Se piensa que el DNA puede pasar de una forma a otra al interactuar con componentes celulares. Se cree que el DNA adopta la forma A cuando está formando híbridos. Con menos del 75% de humedad adopta la forma A.
EL DNA-A presenta 11 nucleótidos por vuelta. Es una hélice más corta y ancha que la del ADN-B. Aquí los pares de bases no aparecen perpendiculares al eje mayor de la hélice como aparecía en la hélice ADN-B. Los surcos mayor y menor están menos diferenciados en tamaño. Es también dextrógira.
El DNA-Z es levógiro, contiene 12 nucleótidos por vuelta. Es una molécula más delgada y solo hay un único y profundo surco. Los grupos fosfato se disponen en zig-zag.
Se ha observado también la formación de triplex, 3 hélices, e icluso cuartetos. Se dan solo en determinadas secuencias, y posibilitan la formación de diferentes puentes de hidrógeno.
El tamaño de las moléculas de ADN es diferente según el organismo del que proceda. Las más pequeñas constan de miles de pares de bases, y las grandes, de millones de pares.
Se suele hablar del tamaño en kilobases (bases).
Forma
Algunas son lineales, como los cromosomas eucariotas. Otras, con los extremos unidos por enlaces fosfo-di-éster, forman un anillo, como los cromosomas de bacterias, ADN mitocondrial, de ciertos virus, y de los plásmidos (ADN en el citoplasma que se replica autónomamente).
Los ADN circulares se superenrrollan sobre sí mismos, y a estos se les denomina ADN's superenrrollados
También pueden asociarse como vitaminas para enrollarse más.
La eschierichia coli, pequeña, tiene un cromosoma de lngitud 1mm, y está superenrrollado y asociado a prtoteínas y a ciertos componentes de la membrana plasmática.
El ADN lineal no puede superenrrollarse. En eucariotas se pliega alrededor de proteínas y así es como se empaqueta para formar la cromatina.
La estructura de la cromatina es muy variable, pudiendo encontrarse desde estados poco compactos (fibras de 10 nm de diámetro) hasta estados muy empaquetados (durante la metarase). Entre estos grados, existen intermedios.
La cromatina está formada por las proteínas “histonas”, ficas en aminoácidos básicos (cargadas positivamente) a pH fisiológico lo que les permite interactuar con los grupos fosfato (cargados negativamente) del DNA.
En células de mamíferos existen 5 tipos: , , , y .
Las cuatro últimas están formando un octámero, con dos unidades de cada una. Alrededor de éste, da dos vueltas la fibra del ADN, formando así los nulceosomas, unidad repetitiva de la fibra de cromatina.
El ADN que une los nucleosomas es el “espaciador”, que se une al siguiente espaciador por una molécula de .
Esta fibra se pliega para dar lugar a una fibra más gruesa, de 30 nm, en la que cada 6 nucleosomas dan una vuelta de hélice (se enrollan). Así se consigue empaquetar la cromatina unas 50 veces.
En la metafase el empaquetamiento es de 100 veces más. Esto se alcanza por interacción con otras proteínas.
El DNA mitocondrial es mucho más sencillo y es circular.
Debido a que las uniones del ADN son puentes de hidrógeno, es posible separar las dos cadenas rompiendo las uniones con un aporte bajo de energía. Esto produce la desnaturalización o fusión del ADN, y puede realizarse por un aumento de pH o de temperatura.
La temperatura a la que se desnaturaliza el ADN al 50% es la o temperatura de fusión, y es una función directa del contenidos de pares citosina-guanina, pues se enlazan por 3 puentes de hidrógeno. A mayor concentración de guanina-citosina mayor temperatura.
Este proceso es reversible. Si volvemos a temperatura ambiente, el dúplex vuelve a formarse, denominándose a esto “renaturalización”
La desnaturalización da lugar al “efecto hipercrómico”, que consiste en que cuando se desnaturaliza, aumenta su absorbancia en el ultravioleta.
Lo que absorbe la UV, en realidad, son las bases nitrogenadas, y cuando se separan las hebras, estas quedan expuestas, captando así mejor la UV.
Existen métodos como la absorbancia para determinar ácidos nucleicos, o como la transformación de estos en compuestos coloreados, que pueden ser determinados por colorímetros. Un coloreador es el “orcinol”, específico para ribosa, o la difenilamina, para la desoxirribosa.
El DNA también se puede identificar por fluorescencia con el bromuro de “eidio”, que se intercala entre bases y emite una coloración característica.
Secuencias palindrómicas
Son aquellas secuencias que se pueden leer igual en un sentido u otro. Por ejemplo:
5'-AAATTT-3' = 3'-TTTAAA-5'
Son regiones con un tipo de simetría binaria importantes porque se forman estructuras secundarias características en los ácidos nucleicos a los cuales se unen proteínasque regulan la expresión genética, pero porque también porque hay encimas de restricción que rompen los enlaces fosfo-di-éster, y algunas reconocen estas secuencias y son dianas de algunas encimas.
ARN
Estructura de los ácidos ribonucleicos
Son moléculas monocatenarias de tamaño mucho más pequeño. Estas tienen entre 70 y 10.000 nucleótidos.
Aunque la molécula es monocatenaria, pueden existir regiones bicatenarias, que se forman por emparejamiento antiparalelo de bases en la misma cadena, formando lazos, orquillas...importantes para la función de ese ARN.
Las cadenas de ARN pueden unirse con cadenas complementarias de ADN, dando lugar a la hibridación.
El RNA es más abundante que el DNA en la célula, y existen diferentes tipos de ARN's:
- ARNm (o mensajero),
- ARNr (ribosómico),
- ARNt (transferente),
- ARNsn (nuclear pequeño),
- ARN heterogéneo nuclear.
El más abundante es el rRNA, y representa el 80% del total de los RNA's en la célula, seguido del transferente.
Además, se han identificado los denominados “ribozimas”, que son RNA's que presentan por sí mismos función catalítica.
El ARNm:
Se caracteriza porque no presenta una configuración típica que lo diferencie de los demás, ya que en una célula puede existir al mismo tiempo miles de moléculas de ARNm con una secuencia o estructura diferente.
El mensajero contiene la información que dirige el ensambjaje de los aminoácidos para formar los polipéptidos. Esta información está contenida en tripletes de bases denominados “codones”, específicos para cada aminoácido.
Los ARN mensajeros de procariotas y eucariotas son distintos. El de eucariotas es monogenico, contiene la información para un único polipéptido, mientras que en procariotas, este es poligénico, y puede codificar varios polipéptidos. También hay diferencias en el sistema del proceso de maduración del ARN.
El ARNt:
Los RNA transferentes son los más pequeños, porque contienen entre 70 y 95 nucleótidos. Existen en las célula vivas muchos diferentes (hasta 60).
La función de estos ácidos nucleicos es unir a los aminoácidos en la biosíntesis de proteínas, siguiendo el mensaje que le da el ARNm. Colaboran en el ensamblaje de los aminoácidos correctamente.
Esto lo sigue por el anticodón, un triplete de pares de bases complementaria al codón del ARNm.
Las moléculas del transferente son monocatenarias, pero existen regiones con apareamientos de bases intracatenarios. Esto se manifiesta porque cuando los transferentes se desnaturalizan, muestran efecto hipercrómico.
La estructura bidimensional, secundaria, de los transferentes es la estructura en “trébol”, que consta de un extremo abierto, tres brazos principales, y un brazo menor. A su vez, este trébol se pliega sobre sí, y a esta estructura se la denomina en “L”.
Todos los transferentes tienen en el brazo abierto los extremos 5' y 3'. Todos acaban con CCA en el 3', que es donde se une el aá. por enlaces covalentes durante la biosíntesis proteica.
El brazo C está asociado con la fijación del aminoacil tRNA (RNA + aá.)
El asa 2 es donde está el anticodón, y se caracteriza por tener en las posiciones adyacentes nucleótidos modificados.
El asa 3 contiene dihidrouracilo. Interviene también en la fijación al ribosoma del aminoacil tRNA.
Una característica de todos los ARNt es la presencia de nucleótidos modificados. Hay una gran variedad. Los más comunes son bases metiladas, nucleótidos con dihirouracilo, y también la “pseudouridina”, representada por “”(fi). Aquí lo que varía frente al nucleótido normal es que la base nitrogenada se une a la ribosa por el y no con el 1, lo normal.
ARNr
El ARN ribosomal es el más abundante. Constituye aproximadamente el 65% de los ribosomas.
Los ribosomas intactos don diferentes en procariotas y eucariotas.
En procariotas, el tamaño es de 70S y está formado por dos subunidades, una mayor (de 50S) y una menor (de 30S). La unidad mayor o “pesada” contiene 2 tipos de RNA, de 23S y de 5S. La menor o ligera, solo posee un tipo de 16S.
En eucariotas el ibosoma es de 80S. La subunidad mayor, de 60S, contiene tres RNA's, de 28S, 5S, y 5,8S, mientras que la pequeña, de 40S, solo tiene un tipo, de 18S.

Secuencia y estructura de los ARNr:
Sus secuencias están muy conservadas. Son muy parecidas en todas las especies en la escala evolutiva. También presentan estructuras secundarias determinadas por la formación de apareamientos de bases nitrogenadas intracatenarias.
Toma formas diversas según la secuencia. Estas estructuras secundarias influyen en la asociación con las proteínas.
ARNsn (small nuclear RNA)
Estos, se llaman así por su pequeño tamaño (entre 4S y 8S) y porque aparecen en el núcleo. Están en eucariotas, procariotas y virus.
Muchos de estos ARN existen en estado libre, y se asocian con proteínas para formar partículas de ribonucleoproteínas.
El más abundante es el U-snRNA, donde la “U” proviene de que es rico en uracilo y se ha encontrado en núcleos de células eucariotas. Se han descrito 6 tipos de estos U-snRNA. Además de ser ricos en U, también se caracterizan porque contienen pseudouridina, bases metiladas, y porque contiene un elemento estructural en el extremo 5' llamado CAP, presente también en RNA mensajeros de células eucariotas. Este casquete 5' consta de un nucleótido de G en el que la guanina está trimetilada, que se une por un enlace trifosfato 5'-5' a 1 ó 2 moléculas de azúcar que están metiladas en posición 2'.
La función que se ha propuesto para los U-snRNA es en el proceso de maduración del RNAm en eucariotas. Estos participan en la escisión (eliminación) de los intrones, aquellas regiones que no se traducen) y en el empalme de los exones (aquellas que sí se traducen).
A algunos ARN se les ha encontrado función catalítica, lo que ha llegado a cuñar el término de ribozima, ARN con función encimática.
1
Descargar
| Enviado por: | El remitente no desea revelar su nombre |
| Idioma: | castellano |
| País: | España |
Todos los derechos reservados.