Psicología
Dopamina y conducta
ÍNDICE
1.- SINAPSIS
2.- DOPAMINA
2.1- Catecolaminas
2.2- Definición
2.3- Metabolismo
2.4- Vías dopaminérgicas
2.5- Receptores dopaminérgicos
2.6- Acciones farmacológicas periféricas
2.7- Agonistas dopaminérgicos
2.8- Antagonistas dopaminérgicos
2.9- Farmacos que actuan sobre la sinapsis dopaminérgica
3.- TRASTORNOS ASOCIADOS A LA DOPAMINA
3.1- ENFERMEDAD DE PARKINSON
3.1.1- Introducción
3.1.2- Características neuropsicológicas
3.1.3- Tratamiento
3.1.4- Pronóstico
3.1.5- Programa de ejercicios
3.2- SINDROME DE TOURETTE
3.2.1- Concepto e historia
3.2.2- Los tics
3.2.3- Ecolalia, ecopraxia, palilalia y otras repeticiones
3.2.4- Problemas emocionales, de comporamiento y otros relacionados
3.2.5- Tratamiento
3.2.6- Herencia y TS
3.2.7- Investigaciones recientes
3.3- ESQUIZOFRENIA
3.3.1- Introducción
3.3.2- Clasificación de la esquizofrenia
3.3.3- Consideraciones neuroanatómicas e inmunohistoquímicas
3.3.4- Consideraciones neuroquímicas
3.3.5- Aminoácidos excitatorios y esquizofrenia
4.- DROGAS RELACIONADAS C CON LA DOPAMINA
1.- SINAPSIS
Se denomina sinapsis al lugar de conexión de dos neuronas. Las
Neuronas utilizan un código digital para conducir información a lo largo del axón, pero para transmitir dicha información a la siguiente neurona se valen de un código analógico de índole química y no eléctrica. A medida que el axón va llegando a su destino se ramifica mucho y en el extremo de cada ramificación se encuentra una pequeña dilatación denominada terminal sináptica; cuando un potencial de acción llega a una de estas terminales sinápticas se libera una pequeña cantidad de una substancia química llamada neurotransmisor; esta sustancia se difunde por el pequeño espacio existente entre la terminal sináptica y la membrana de la siguiente célula (este espacio se denomina corredor sináptico y puede medir de 120 a 200 unidades ängstrom, y por lo mismo no es una distancia muy grande que deban cruzar las moléculas de neurotransmisor). Al llegar el neurotransmisor a la célula vecina produce un cambio en su membrana que a su vez provoca una hiperpolarización o una despolarización de pequeña intensidad. Estas últimas se conducen al cono axónico en donde se integran con los mensajes que están llegando a la neurona procedentes de otros sitios de las dendritas y del cuerpo celular. De ordinario, la información pasa de la célula presináptica, por la sinapsis, a sitios de las dendritas de la célula postsináptica; este tipo de sinapsis recibe el nombre de sinapsis axo-dendrítica, porque en ella intervienen las terminaciones axónicas de una célula y las dendritas de la siguiente; siendo esta la más habitual, existen otra serie de contactos como las sinapsis axo-somáticas, las axo-axónicas e incluso las dendro-dendriticas, que como su nombre indica intervienen en ella las dendritas de dos neuronas. Además existen axones que tienen como célula postsináptica a células musculares, vasos sanguíneos, glándulas endocrinas, etc.
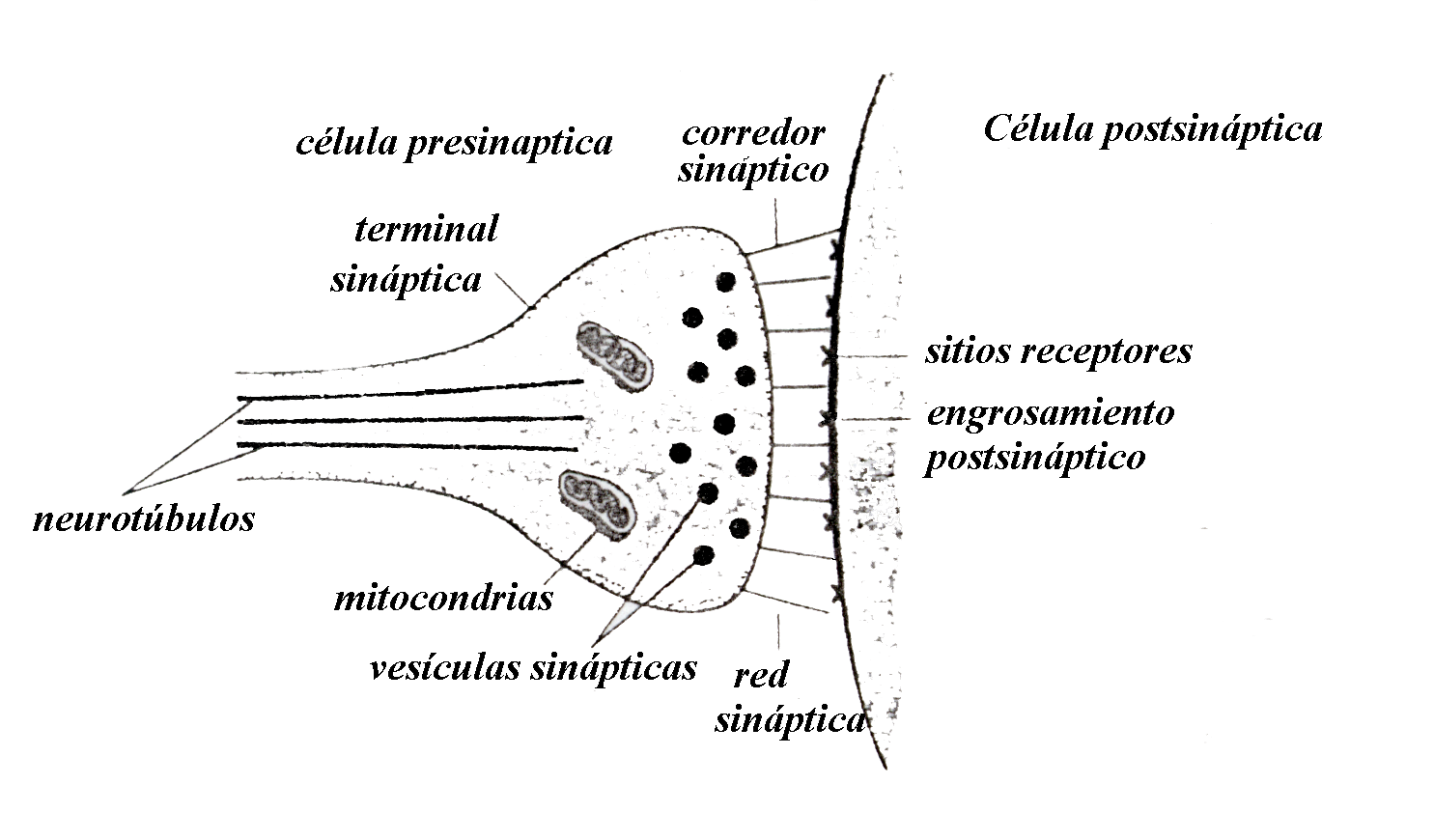
La transmisión sináptica en los contactos axo-dendríticos y axo-somáticos
comprende tres fenómenos básicos:
- Liberación del neurotransmisor: Cuando el potencial de acción despolarizante pasa por el axón y luego por todas sus ramas, finalmente produce una rápida despolarización de la membrana en las terminales sinápticas. A causa de esta despolarización, una o más de estas vesículas liberan su contenido en el corredor sináptico. La cantidad liberada de neurotransmisor dependerá del cambio rápido de potencial de membrana desde el nivel de reposo (-70 Mb) hasta el que alcanza en el punto máximo del potencial de acción (+50 mV). Por lo general como todos los potenciales de acción tienen la misma intensidad, la cantidad de neurotransmisor que se libera es igual en cada punto máximo o acmé (sin embargo en los contactos axo-axónicos puede variar la cantidad de neurotransmisor liberada).
- Potenciales postsinápticos: Ya que las moléculas de neurotransmisor se han vaciado en el corredor sinápticos, se difunden a través de éste y ejercen su efecto en la membrana postsináptica. Cuando llegan allí, se unen muy brevemente a los sitios receptores de la membrana postsináptica. La permeabilidad de ésta se altera ligeramente cuando el neurotransmisor se une y cambia la concentración de iones del interior y del exterior de la membrana. Si se tomara un registro de la célula postsináptica inmediatamente después de que se ha liberado el neurotransmisor, observaríamos dos fenómenos: una despolarización o una hiperpolarización seriada; que corresponden a los llamados potenciales de acción postsinápticos o PSP en abreviatura. Cuando el PSP es una despolarización se le da el nombre de potencial postsináptico excitador (EPSP), ya que tiende a aumentar la probabilidad de que en el cono axónico se dispare un potencial de acción. Un PSP que se produce durante un hiperpolarización recibe el nombre de potencial postsináptico inhibidor, dado que tiende a inhibir el disparo de un potencial de acción cuando llega al cono axónico.
Depende de dos hechos que un neurotransmisor liberado produzca un EPSP o un IPSP en la célula postsináptica. Primero, el tipo de sustancia neurotransmisora que use la célula presináptica. Segundo, la naturaleza del tipo receptor. Así un mismo neurotransmisor puede provocar un EPSP, o un IPSP, según la sinapsis y el tipo de sitio receptor con que se combine. Estos PSP no son como los potenciales de acción; no provocan cambios súbitos en el potencial de membrana. Se trata de modificaciones graduales que rápidamente se propagan al cono axónico y se integran a los demás influjos que la neurona está recibiendo. Es importante recordar la diferencia entre estos potenciales seriados y el tipo de espigas (acmés) todo o nada que se desarrollan en el axón. No obstante, un EPSP gradual puede desencadenar un potencial de acción si tiene la suficiente magnitud para alcanzar el umbral y si llega en ese momento al cono axónico, muy pocos IPSP que podrían cancelar al potencial de acción. Aún no se conoce con claridad la naturaleza exacta de los sitios receptores y los efectos que en ellos ejerce la substancia neurotransmisora. Quizá los sitios receptores actúen como puertas en la membrana que controla el flujo de iones: el neurotransmisor tendría entonces la capacidad de abrir algunas de las puertas.
Movimientos iónicos durante el EPSP. Cuando un neurotransmisor produce un EPSP en la célula postsináptica aumenta la concentración de iones positivos en el interior de ésta. Eccles supone que un EPSP se debe principalmente a influjo de iones de sodio. Cuando el neurotransmisor se combina con los sitios receptores, los poros de la membrana se abren un poco más y permiten el paso de algunos iones de sodio hacia la célula. Esto haría al interior ligeramente más positivo y explicaría la despolarización que ocurre durante un EPSP. Un efecto de esta hipótesis es que todo poro lo suficientemente amplio para admitir sodio tiene que serlo también para que por él pasen iones de potasio y cloro. Eccles ha propuesto que algo de potasio sale de la célula durante un EPSP. Si no fuera así, el EPSP sería aun más grande. Sin embargo, los iones de cloro de carga negativa, que tenderían a desplazarse al interior de la célula junto con los de sodio por su gradiente osmótico, no lo hacen porque los poros se revisten de cargas negativas; éstas repelerían a los iones de cloro de carga negativa pero atraerían a los de sodio y potasio, de carga positiva.
Movimientos de los iones durante un IPSP. Cuando un neurotransmisor provoca un IPSP, encontramos que el interior de la célula se hace más negativo que el exterior. Este fenómeno se debe probablemente a que los poros se abren lo suficiente para permitir el paso de iones de potasio y cloro, pero demasiado pequeña para que pase el sodio. Cuando el neurotransmisor se combina con los sitios receptores, pequeños poros permiten que escape el potasio (K+), siguiendo el gradiente osmótico. También salen iones de cloro porque siguen su gradiente electrostático. La perdida neta de potasio daría lugar a despolarización.
Los EPSP y los IPSP que ocurren en la célula postsináptica cuando se libera el neurotransmisor de la célula presináptica atraviesan el cuerpo celular y llegan rápidamente al cono axónico. Allí se integran y el cono axónico dispara un potencial de acción cuando la integración ha dado lugar a una despolarización neta que pudo alcanzar el umbral axónico. Una sola neurona puede estar recibiendo influjo en la forma de IPSP y de EPSP de muchas sinapsis que están esparcidas por las dendritas y el cuerpo celular. Todos se integran mediante los procesos de sumación espacial y temporal.
- Inactivación del neurotransmisor: Una vez liberado en neurotransmisor, por la terminal sináptica, es muy importante que sólo tenga un efecto muy breve en la membrana postsináptica. Las moléculas de neurotransmisor no deben quedar permanentemente unidas a los sitios receptores, ya que esto haría imposible la recepción de nuevos mensajes. Existen dos mecanismos para asegurar la brevedad del efecto del neurotransmisor en la membrana: inactivación por enzimas y recaptación.
Inactivación por enzimas. Las enzimas se encuentran en la membrana postsináptica, de modo que tan pronto como llega un neurotransmisor es desdoblado por una enzima; entonces los subproductos ya no pueden interactuar con los sitios receptores. Un ejemplo de esto es la acetilcolinesterasa que inactiva al neurotransmisor acetilcolina.
Recaptación. En lugar de ser incativados por enzimas de la membrana postsináptica, algunos neurotransmisores son llevados directamente de regreso a la membrana. Tan pronto como se libera el neurotransmisor, la terminal sináptica lo recaptura, de modo que sólo tiene un breve lapso para ejercer su efecto en la membrana postsináptica. Las sinapsis que usan el neurotransmisor adrenalina, por ejemplo, utilizan este mecanismo de recaptación para interrumpir los efectos de las sustancia en la siguiente célula.
2.- DOPAMINA
2.1- Catecolaminas
Las monoaminas constituyen el grupo principal de neurotransmisores del sistema nervioso. La característica diferencial de estas sustancias es la presencia de un grupo amino (-NH2), por lo que se denominan monoaminas o también aminas biogénicas. Proceden de aminoácidos precursores y forman dos grupos: las catecolaminas, derivadas de la fenilalanina; y las indolaminas, que derivan del triptofano.
Las catecolaminas incluyen la dopamina, la noradrenalina y la adrenalina; mientras que en las indolaminas es la serotonina su neurotransmisor. La diferencia entres ellas hace referencia al núcleo o anillo central de su estructura molecular como se corresponde con la propia diferencia estructural.
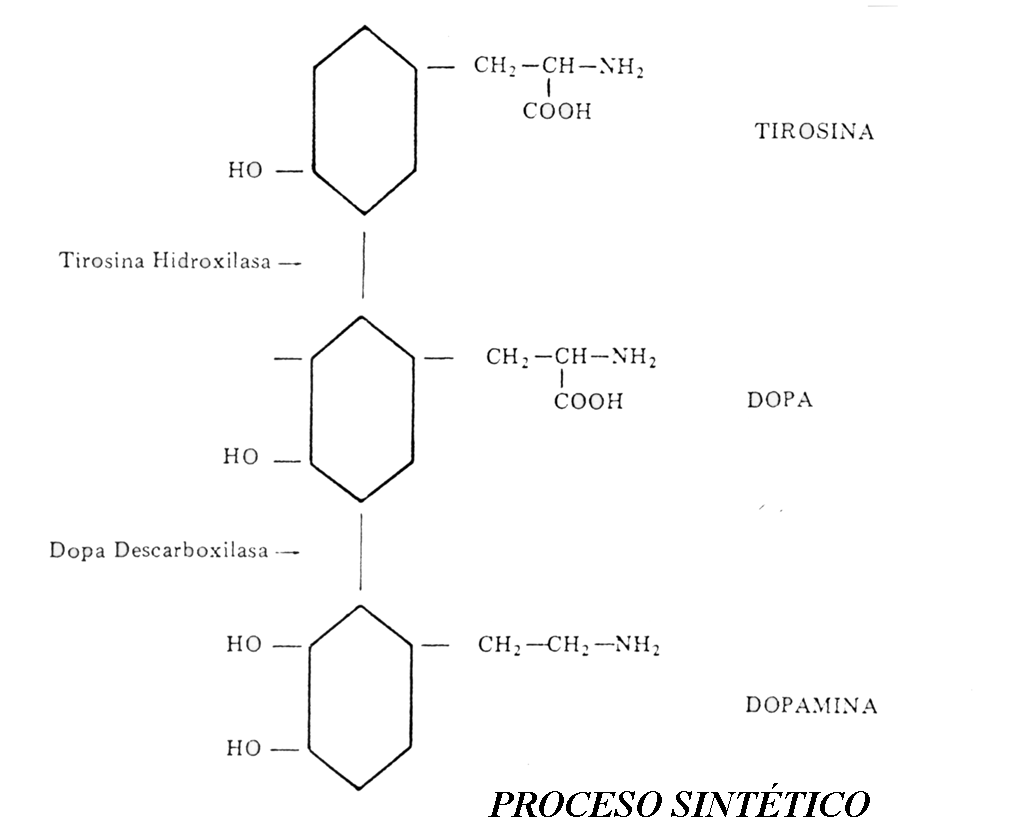
La síntesis de las distintas catecolaminas se da en fases sucesivas del proceso metabólico e interviniendo sistemas enzimáticos adicionales. Todas las catecolaminas proceden del aminoácido Fenilalanina, que se transforma en tirosina en el hígado. Desde éste la tirosina es vertida al torrente sanguíneo. La enzima que cataliza esta reacción es la Fenilalanin Hidroxilasa. La tirosina que circula por la sangre pasa al del vaso sanguíneo al interior de la terminación nerviosa. Aquí se da el paso siguiente, la transformación de la tirosina en Dopa o dihidroxifenilanina. La enzima reguladora es la Tirosin-hidroxilasa. Esta enzima es la de mayor especificidad y la que existe en menor cantidad de todas cuantas intervienen en el proceso sintético, por lo que se puede decir que es el principal regulador del proceso (factor limitante de la reacción).
El paso siguiente consiste en la transformación de la Dopa en Dopamina (primera catecolamina). La enzima que interviene es la Dopa-descarboxilasa (L-aminoácido-descarboxilasa), de acción poco específica. El paso de Dopamina a Norepinefrina, regulado por la Dopamina-beta-hidroxilasa, no tiene lugar como los anteriores, en la matriz citoplasmática, sino dentro de la vesícula sináptica, donde queda almacenada la NE. La transformación de Ne a epinefrina es regulada por la fenil-etanolamina-N-metil-transferasa (PNMT). La epinefrina ejerce también un efecto inhibidor sobre esta enzima, tratándose de otro mecanismo de feedback negativo.
2.2- Definición
La catecolamina dopamina es una sustancia neurotransmisora que se encuentra en el cerebro cuyas redes neuronales intervienen en gran parte de la regulación del movimiento. La dopamina es la base de numerosos trastornos tales como la esquizofrenia, la enfermedad de Parkinson, la depresión, el síndrome de Tourette, etc (dedicaremos un capitulo a los trastornos asociados a la dopamina). Las sinapsis que emplean la dopamina reciben el nombre de sinapsis dopaminérgicas. Como ya hemos dicho la dopamina pertenece al grupo de las catecolaminas, en la síntesis de las mismas es un paso previo a la Nor-epinefrina (o Nor-adrenalina). Precisamente fue la primera catecolamina de la que se demostró su existencia en algunas áreas cerebrales, con lo que se consiguió uno de los primeros indicadores de la existencia de neuronas catecolaminérgicas en el Sistema Nervioso Central.
En fechas bastante recientes, la década de los 60, se consiguió poner de manifiesto aquellas sinapsis que tienen DA como neurotransmisor, mediante técnicas histoquímicas de fluorescencia. Esta técnica consiste en convertir el transmisor natural en un derivado fluorescente que brillará al ser expuesto a la radiación ultravioleta en el microscopio de fluorescencia.
La DA está distribuido irregularmente en el cerebro y no guarda relación su presencia, en mayor o menor grado, con la riqueza de vasos sanguíneos; este hecho fue precisamente el que hizo sospechar en su función como neurotransmisor, lo que posteriormente se demostró.
Podemos decir que la dopamina es la catecolamina más importante precisamente porque presenta una localización encefálica más elevada que la noradrenalina y, por tanto, su repercusión comportamental es más comprometida. Los núcleos cuneiformes, el núcleo rojo, la sustancia negra y las áreas tegmentales son estructuras claramente dopaminérgicas. Lo más significativo de estas estructuras, es que sus fibras constituyen tres fascículos altamente característicos del encéfalo: el fascículo nigroestriado, que se proyecta esencialmente a los núcleos grises basales; el fascículo mesocortical, que alcanzan además de estructuras internas de la corteza, la gran estructura prefrontal. De ahí que la acción de la dopamina debe estar inexorablemente implicada en el control y regulación del movimiento, en la expresión de los estados afectivos y en la capacidad de proposición y juicio, como anteriormente hemos referido.
Hay, asimismo, presencia dopaminérgica a nivel talámico e hipotalámico, de una manera menos concreta, por lo que se habla de proyecciones inespecíficas que se proyectan sobre las estructuras hipotálamo-hipofisarias y tienen una clara repercusión en la regulación de la economía y el metabolismo orgánico a través de la secreción de factores de liberación de las hormonas hipofisarias.
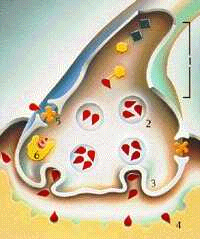
Esquema de los procesos de la dopamina
1.- Producción de dopamina
2.- Almacenamiento de dopamina
3.- Liberación de dopamina
4.- Receptores para la dopamina
5.- Recaptación de dopamina
6.- Eliminación de la dopamina
2.3- Metabolismo
Síntesis, almacenamiento y liberación.
El proceso sintético es el mismo que de la norepinefrina. El aminoácido Fenilalanina, normalmente es transformado en Tirosina en el hígado, por acción de la Fenilalanina-Hidroxilasa. La transformación de la Tirosina en Dihidroxifenilalanina (DOPA), está regulada por la enzima Tirosin-hidroxilasa y tiene lugar ya en la terminación nerviosa. La DOPA es descarboxilada por la enzima Dopa-descarboxilasa, con lo que aparece la DOPAMINA, ésta se almacena en el pie terminal del axón, en unos minúsculos sacos delimitados por una membrana llamados vesículas sinápticas. En un solo pie terminal hay millares de ellas y casa una contiene entre 10.000 y 100.000 moléculas de DA. La llegada de un impulso nervioso al pie del axón es la causa de la liberación, que se desencadena porque aumenta la permeabilidad del pie terminal a los iones de calcio, los cuales activan los mecanismos de liberación. El mecanismo no es bien conocido.
b) Destrucción
Una forma de destrucción es la posible transformación de la DA, en NE. Ahora bien, existen también vías metabólicas enzimáticas a cargo de la MAO y la COMT, las cuales pueden actuar. LA Dopamina (DA), por acción de la COMT que se encuentra preferentemente a nivel del espacio intersináptico, es transformada en 3-Metoxitiramina (3MT). La 3MT por acción de la MAO extraneuronal, es transformada en Ácido Homovanílico(HVA).
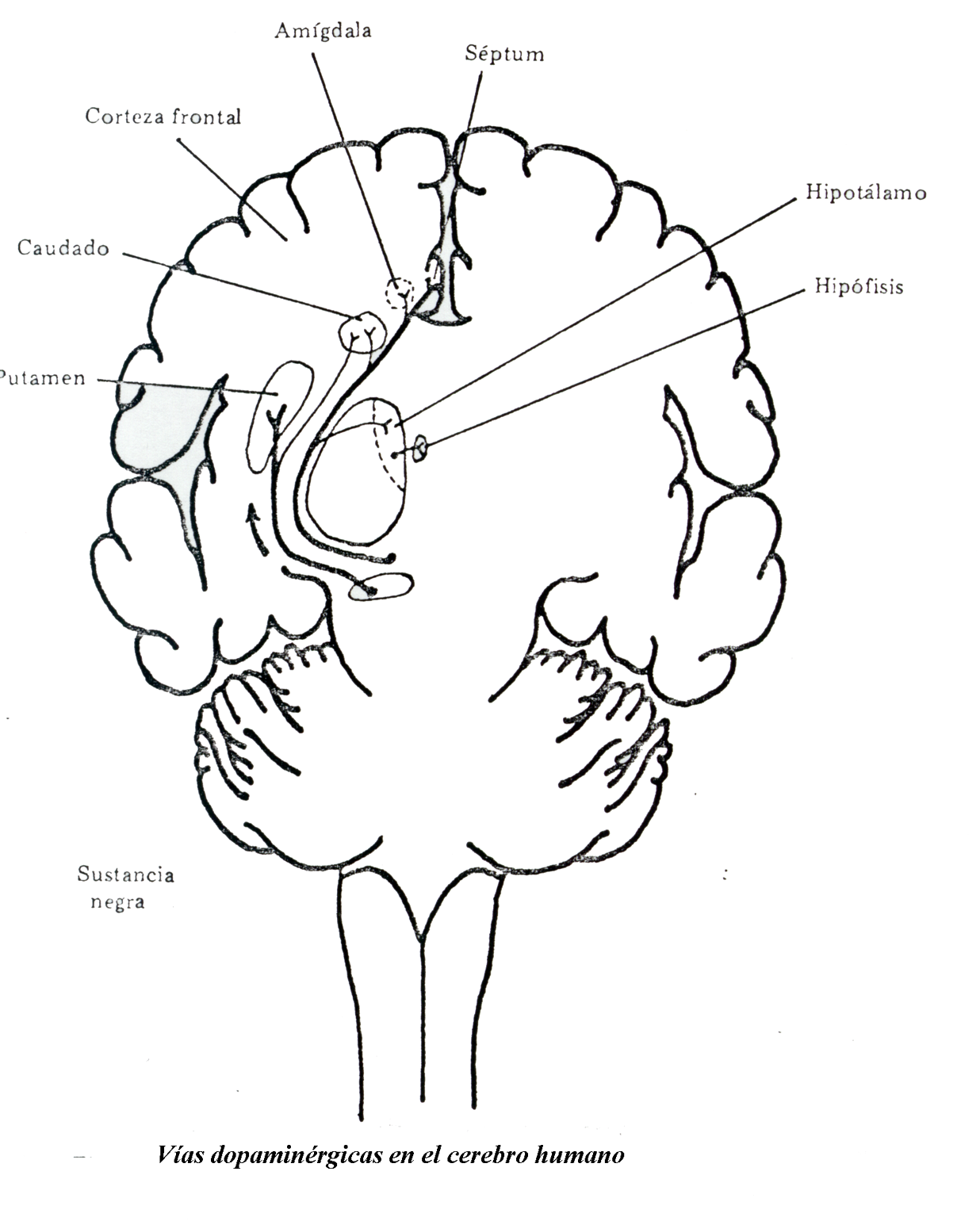
2.4- Vías dopaminérgicas
El sistema dopaminérgico tiene una distribución más restringida y una función mas definida que el sistema noradrenérgico. En la actualidad se conocen cuatro sistemas dopaminérgicos bien definidos, que son los siguientes:
a) Sistema Nigroestrial
Se origina en la zona compacta de la Substancia Negra (pequeña región del mesencéfalo), y desde allí se proyecta al Cuerpo Estriado, mediante fibras muy ramificadas.
b) Sistema Tuberoinfundibular
Tiene su origen, principalmente, en el Núcleo Arcuatus del Hipotálamo e inerva la capa externa de la eminencia media y la porción nerviosa e intermedia de la Hipófisis.
Una característica de este sistema es que cuando sus neuronas se activan durante un tiempo largo, se depeccionan marcadamente de DA, y que no son destruidas por la 6-hidroxidopamina (substancia muy tóxica que se forma por hidroxilación del carbono 6 de la DA, en el espacio intersináptico).
c) Sistema Mesolímbico
Las células que forman esta vía tienen su origen en el mesencéfalo. Fueron denominadas neuronas A 10 por Dahltrom y Fuxe (1964). Las áreas inervadas por estas células incluyen los Tubérculos Olfatorios y los Núcleos Accumbeus; los Núcleos centrales de la Amígdala y el Núcleo Lateral del Septum. Evidencias recientes sugieren que las neuronas A 10 se proyectan también a los núcleos basales laterales y posteriores laterales de la amígdala, así como al núcleo caudado ventral lateral.
d) Sistema Mesocortical
No es todavía bien conocido si las células A10 tienen ramas que inervan, unas las áreas límbicas y otras las áreas corticales, o si son células independientes para cada sistema. En términos de respuesta a las drogas las neuronas A10 aparecen relativamente homogéneas.
En 1973 se sugirió la existencia de una proyección dopaminérgica cortical, basándose en evidencias bioquímicas. También en ese mismo año se descubrió una adenil-ciclasa sensible a la DA, en el cortex frontal.
Concretamente la inervación dopaminérgica puede ser detectada en el Cortex Prefrontal, Circunvolución del Cíngulo y Cortex Entorrinal.
2.5- Receptores dopaminérgicos
Las moléculas de DA liberadas cruzan velozmente el espacio intersináptico actuando sobre unos lugares receptores específicos situados en la membrana postsináptica. Estos receptores están constituidos por moléculas de proteínas. Existe en la superficie de la proteína receptora una región que corresponde exactamente con la forma y configuración de la molécula de Dopamina. Además de este “primer receptor”, se ha demostrado que la DA actúa sobre una sustancia que hace a modo de “segundo mensajero”. SUTHERLAND identifico la sustancia de “segundo mensajero” como la molécula de AMP cíclico. Según este autor, la proteína receptora de la DA está acoplada al enzima adenilciclasa que cataliza la conversión del ATP en AMP cíclico. Seguidamente el AMP cíclico actúa sobre la maquinaria bioquímica de la célula, iniciando la respuesta fisiológica característica del transmisor. La posibilidad de que esta adenilciclasa catalizadora del Amo cíclico, sensible a la dopamina, forma parte del receptor dopaminérgico, viene apoyada por el hecho de que en todas las regiones cerebrales donde la dopamina ha sido localizada como neurotransmisor, se ha descubierto la actividad enzimática de esta adenilciclasa.
Los receptores de dopamina que se conocen hasta hoy son:
- D1: acoplados a la actividad de la adeniciclasa y localizados en la corteza de seres humanos, su activación induce relajación de la fibra muscular lisa en los vasos arteriales renales y de otros territorios vasculares (p. Ej. de musculatura esquelética). A nivel del túbulo renal inhibe la reabsorción de Na+ y aumenta la diuresis. Activa el aparato yuxtaglomerular favoreciendo la secreción de renina.
- D2: asociación negativa a la adenilciclasa. Se reconocen el D2a y el D2b, y se localizan preferencialmente en el sistema límbico y estriado. Al inhibir la función los nervios noradrenérgicos, bloquea la actividad simpática en el corazón y en algunos territorios vasculares; como consecuencia aparece bradicardia y reducción de la resistencia periférica. Este efecto ha sido ampliamente estudiado con fines terapéuticos para controlar la hipertensión o para aliviar la insuficiencia cardiaca congestiva.
- D3: de localización mesolímbica;
- D4: en asociación con el receptor del nucleótido Guanina, y de amplia distribución mesolímbica. Se han identificado las variaciones polimórficas D4-2, D4-4 y D4-7, y es muy probable la existencia de las variaciones D4-6 y D4-8. Este receptor D4 tiene gran afinidad con el antipsicótico clozapina.
- D5: de localización mesocortical y de gran afinidad dopaminérgica, con activación de la enzima adenilciclasa.
2.6- Acciones farmacológicas periféricas
Las acciones de la dopamina son complejas de analizar porque se comporta como activador de baja afinidad de receptores alfa y beta-adrenérgios, y como activador de receptores dopaminérgicos. Además, la estimulación de receptores DA2 presinápticos puede originar una inhibición indirecta de la actividad simpática. Por todo ello la acción resultante es variable y muy dependiente de la dosis, vía de administración y especie animal en que se estudie.
Por su acción adrenérgica produce taquicardia, aumento de la contractilidad cardiaca, arritmias, vasoconstricción y vasodilatación de diversos territorios vasculares.
A nivel de ganglios simpáticos la dopamina inhibe o modera la transmisión ganglionar, aunque la activación de algunos receptores presenta un componente facilitador. En el tracto gastrointestinal produce efectos excitadores e inhibidores tanto sobre la actividad del músculo liso como sobre la secreción exocrina. Los efectos inhibidores de la motilidad (relajación o inhibición de la contracción espontánea) son mas apreciables en el tercio inferior del esófago, estómago, intestino delgado y grueso; pero como antes se ha indicado, no está claro si se debe a activación de receptores dopaminérgicos. En cuanto a la actividad secretora en la especie humana, la acción de la dopamina es bastante dudosa.
2.7- Agonistas dopaminérgicos
Pertenecen a muy diversas clases químicas, existiendo una complejísima relación entre estructura y actividad, según el tipo de receptor considerado, el modelo farmacológico en que se estudie, las condiciones experimentales, etc. Muchos de ellos contienen en su molécula el esqueleto fundamental de la estructura dopamínica
Los grupos generales son los siguientes:
Feniletilaminas. Son la dopamina (DA) y sus derivados directos por sustitución en el átomo de nitrógeno, en la cadena lateral, o en el anillo: epinina (N-metil-DA); derivados N-sustituidos
Aminotetralinas y ainoindanos. Destacan la 5,6-dihidroxi-2-aminotetralina (5,6-ADTN) y la 6,7-ADTN y otros derivados
Derivados ergóticos. Tienen acción agonista pura o parcial, según el tipo de receptor. Se encuentran los derivados peptídicos (p. Ej., bromocriptina) y los derivados ergolénicos (p. Ej., pergolida, lisurida, elimoclavina)
Octahidrobenzoquinolinas
Aporfinas. Destacan la apomorfina y la N-propilnorapomorfina
Estructura variada: benzacepinas, piribedilo, indolonas.
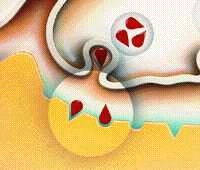
En la práctica, no se encuentran agonistas plenamente selectivos de un tipo u otro de receptor. Pero los derivados ergóticos son agonistas preferentes de los receptores D2 y DA2.
Los agonistas de la Dopamina trabajan estimulando directamente receptores de la dopamina La Dopamina trabaja principalmente sobre dos receptores: Dl y D2 Los agonistas de la Dopamina trabajan sobre uno o varios de estos receptores: Dl y D2
2.8- Antagonistas dopaminérgicos
Las principales familias pertenecen a los grupos de fármacos denominados antipsicóticos. Las fenotiacinas y tioxantenos son antagonistas inespecíficos, las benzamidas bloquean más selectivamente los receptores DA2 y D2, las buitrofenonas ocupan una posición intermedia, y el nuevo fármaco SCH 23390 se comporta como antagonista DA1 y D1.
2.9 - Fármacos que actúan sobre la sinapsis dopaminérgica
Las sustancias que alteran la síntesis de NE también lo hacen con la de dopamina, ya que participan en la misma vía biosintética. Sin embargo, algunas otras afectan las sinapsis dopaminérgicas de forma selectiva.
De los fármacos que actúan sobre la sinapsis dopaminérgicas destacan los siguientes:
L-DOPA.- Favorece la síntesis de la Dopamina, consiguiendo un aumento de la misma en el espacio sináptico.
RESERPINA.- Inhibe la liberación de Dopamina, haciendo menor su concentración. La Reserina produce en el hombre los síntomas característicos de la enfermedad de Parkinson. Se ha visto que en esta enfermedad a concentración de DA esta alterada, existe una clara deficiencia en los ganglios basales. También se ha detectado una disminución de HVA, principal metabolito de la DA.
ANFETAMINA.- Estimula la liberación de Dopamina, puede llegar a producir una sintomatología psicótica similar a la esquizofrenia paranoide. En este sentido se ha asociado un exceso de DA en la zona límbica del encéfalo anterior con la esquizofrenia.
APOMORFINA.- Activa la liberación de Dopamina y estimula los receptores dopaminérgicos.
BENZOTROPINA.- Inhibe selectivamente la recaptación de DA en las terminales sinápticas, aumentando el tiempo de permanencia de ésta en la sinapsis. Esta substancia aumenta la cantidad de dopamina en la sinapsis y por lo mismo potencia los efectos de ésta en la membrana postsináptica. Por lo que se administra en enfermedades en que las sinapsis dopaminérgicas están dañadas, por ejemplo la enfermedad de Parkinson.
PIMOCIDE.- Bloquea los receptores dopaminérgicos.
NEUROLÉPTICOS.- Tales como la Clorpormezina y el Haloperidol. Bloquean los receptores dopaminérgicos. Tienen poderosos efectos antipsicóticos, hecho que apoya la hipótesis de la relación entre DA y esquizofrenia.
3.- TRASTORNOS ASOCIADOS A LA DOPAMINA
Vamos a tratar en este punto tres de las trastornos mas importantes relacionados con el neurotransmisor dopamina, estos son la enfermedad de Parkinson, el síndrome de Tourette y la esquizofrenia.
3.1- ENFERMEDAD DE PARKINSON
3.1.1- INTRODUCCIÓN
Es una enfermedad neurológica que se asocia a rigidez muscular, dificultades para andar, temblor y alteraciones en la coordinación de movimientos. Es una enfermedad muy frecuente que afecta a 2 de cada 1000 personas, y se desarrolla más a partir de los 50 años, de igual forma a hombres y mujeres.
La enfermedad de Parkinson es un proceso neurológico crónico cuyas causas son :
-
Alteración progresiva en la sustancia nigra del mesencéfalo (ganglios basales y área extrapiramidal). Estas áreas son zonas nerviosas que controlan y coordinan los movimientos.
-
Disminución de la dopamina cerebral. La dopamina es un sustancia neurotrasmisora, que trasmite impulsos de unas células nerviosas a otras.
-
Una lesión anatómica: Se produce un daño progresivo en la sustancia nigra del mesencéfalo (también se afectan otras zonas nerviosas que normalmente controlan y coordinan los movimientos).
-
Un defecto bioquímico: disminuye la dopamina cerebral (también disminuyen otros neurotransmisores).
-
Tres síntomas motores: el enfermo presenta rigidez, temblor e hipocinesia (también se producen otros síntomas).
Actividad Cerebral Normal - Neurotransmisores en equilibrio
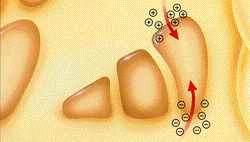
En el cerebro normal la dopamina(-), el principal neurotransmisor que transmite mensajes sobre el movimiento, está en equilibrio con la acetilcolina (+), otro importante neurotransmisor que interviene en el movimiento.
Actividad Cerebral en la Enfermedad del Parkinson: La Pérdida de Dopamina Causa Desequilibrio en los Neurotransmisores
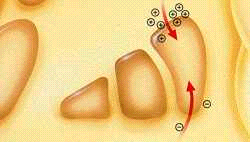
En los pacientes con al enfermedad del Parkinson el deterioro de las células nerviosas comporta la pérdida de dopamina y un desequilibrio de los neurotransmisores [dopamina (-) y acetilcolina(-)] afectando al movimiento.
SINTOMAS
La enfermedad de Parkinson tiene unos síntomas muy característicos:
-
Rigidez muscular.
-
Temblor, puede ser de diferentes intensidades.
-
Hipocinesia, falta de movimientos.
-
Dificultades al andar, parece que se siguen a sí mismos.
-
Mala estabilidad al estar parado, parece que pendulan.
-
Al comenzar a andar tienen problemas, les cuesta empezar.
-
Si un movimiento no se termina tiene dificultades para reiniciarlo, o terminarlo.
-
Cara de pez o máscara, por falta de expresión de los músculos de la cara.
-
Lentitud de movimientos.
-
Acatisia, se dice de una falta de capacidad de estar sentado sin moverse.
-
Movimiento de los dedos como si estuvieran contando dinero.
-
Boca abierta, con dificultad para mantenerla cerrada.
-
Voz de tono bajo, y monótona.
-
Dificultad para escribir, para comer, o para movimientos finos.
-
Deterioro intelectual, a veces.
-
Estreñimiento
-
Depresión, ansiedad, atrofia muscular.
DIAGNOSTICO
El contexto de síntomas es muy característico, y para confirmar el diagnóstico se realiza un TAC que suele presentar lesiones típicas en el mesencéfalo.
Se debe tener en cuenta que la osteoporosis puede producir actitudes musculares similares al Parkinson. Por lo que el examen de los reflejos osteotendinosos, muy exagerados en el Parkinson, serán imprescindibles para el diagnóstico.
3.1.2- CARACTERÍSTICAS NEUROPSICOLÓGICAS
Los primeros descubrimientos acerca de la Enfermedad de Parkinson (EP) estuvieron encaminados a describir principalmente los desórdenes de movimiento. En su descripción original, James Parkinson no consideró los cambios mentales como un síntoma propio de la enfermedad. La primera descripción de estos cambios se le atribuye a Charcot y Vulpian en 1891: "En un momento dado la mente se nubla y la memoria se pierde", enfatizaba Charcot . Posteriormente se incrementaron el número de datos en los que se describían desde alteraciones cognoscitivas específicas hasta cuadros complejos que han sido descritos como síndromes demenciales.
Investigaciones en las últimas décadas han revelado que los trastornos cognoscitivos forman parte de la sintomatología clínica de la EP. La extensa literatura que ha seguido a estos hallazgos se ha enfocado en tres aspectos aun controversiales, el primero se relaciona con las bases patofisiológicas de los trastornos cognoscitivos. Diversas argumentos señalan que alteraciones en los circuitos dopaminérgicos pueden contribuir a la manifestación de los diversos trastornos cognoscitivos, sin embargo en algunos casos se ha encontrado que la perdida dopaminérgica se acompaña de deficiencias colinérgicas y en otros neurotransmisores y estas combinación de anormalidades neuroquímicas exagera los trastornos intelectuales. El segundo aspecto controversial es si la demencia que se observa en algunos pacientes, se debe a la EP o a una demencia senil de tipo Alzheimer concomitante; y un tercer problema se relaciona con la identificación de los mecanismos o procesos que subyacen a los trastornos cognoscitivos específicos que presentan los pacientes con EP que no están demenciados.
Vamos a tratar el punto de las bases patofisiológicas de los trastornos cognoscitivos, posteriormente abordaremos dos de los aspectos mas controversiales que predominan actualmente en el estudio de la neuropsicología del Parkinson. El primer punto que se discute es en relación a la demencia. Se ha postulado que en pacientes con EP existe una demencia de tipo subcortical que es diferente de la demencia senil de tipo Alzheimer (DSTA) .Este concepto de demencia subcortical se sustenta en la base de que existe una correlación entre los déficits cognoscitivos y la disfunción motora, la cual a su vez se atribuye a la pérdida de células dopaminérgicas en los ganglios basales. El segundo punto que discutiremos es el perfil cognoscitivo de los paciente con EP que no presentan demencia, y si estos déficits se deben a una patología subcortical o a una destrucción de fibras eferentes a la corteza. La investigación neuropsicológica ha revelado que existen alteraciones en funciones visoespaciales, especialmente en orientación espacial, y en funciones de ejecución, principalmente en las que se requiere cambiar de set mental . Una de las preguntas que se han originado de estos estudios es si los pacientes con EP tienen un déficit específico visoespacial o si estas dificultades visoespaciales se deben a un déficit más general, por ejemplo una incapacidad para cambiar el set mental . Se han observado también trastornos de memoria especialmente en evocación . En cuanto al lenguaje, las investigaciones reportan dificultades para generar palabras. También existen evidencias experimentales que algunos pacientes con EP tienen problemas afectivos, los cuales pueden manifestarse en síntomas depresivos.
PATOFISIOLOGIA
Se ha demostrado que en la EP existe pérdida neuronal y de pigmentación en la substancia nigra y en otros núcleos subcorticales pigmentados (por ejemplo el locus ceruleus). La severidad de los cambios de la substancia nigra es paralela a la reducción de dopamina en el estriado. Dado que la zona compacta de la substancia nigra contiene la mayor parte de los cuerpos dopaminérgicos del cerebro, estas observaciones sugieren que la vía dopaminérgica nigroestriatal se encuentra lesionada en la EP.
La dopamina normalmente se sintetiza en el estriado, en las terminaciones nerviosas de las neuronas dopaminérgicas cuyos cuerpos celulares se encuentran en la substancia nigra; en estas terminaciones nerviosas se toma el neurotransmisor dentro de las vesículas y se libera en el espacio sináptico cuando las células disparan.
Neuroanatómicamente, la denervación del estriado afecta la salida del estriado a la corteza vía los sistemas estriado-palido-tálamo-cortical (corteza motora) y el sistema nigro (pars reticular) tálamo-cortical (corteza premotora y prefrontal). Se ha propuesto el concepto de circuitos "motor" y "complejo" con respecto a la relación entre ganglios basales y lóbulos frontales.
El circuito motor se dedica al control de los parámetros de movimiento e incluye al área sensoriomotora agranular y áreas corticales premotoras, al putamen, la porción caudal del sistema eferente de los ganglios basales y a una vía de relevo diencefálica vía el núcleo ventral lateral , al área motora suplementaria . El circuito "complejo" tiene una entrada topográficamente organizada de todas las áreas corticales de asociación al núcleo caudado. Transmite información a la porción rostral del sistema eferente de los ganglios basales, con relevo diencefálico vía el núcleo ventral anterior y dorsomedial a los campos frontales oculares y a áreas frontales de asociación que están involucradas en operaciones cognoscitivas.
En la EP, el temblor, la rigidez y la bradiquinesia se deben a la disminución de actividad dopaminérgica en el putamen: "circuito motor". Se ha especulado que la pérdida de fibras pálido-corticales son importantes en la génesis del temblor y que la rigidez se relaciona con la pérdida de fibras estriado-palidales; todo ello está basado en autopsias realizadas en pacientes con EP en los que predominaban uno u otro de estos síntomas. Los síntomas de aquinesia y los defectos posturales y del equilibrio son síntomas derivados de la degeneración de cuerpos celulares en la substancia nigra.
La patofisiología de los trastornos cognoscitivos en la EP aún es controversial. Sin embargo diversos factores sugieren que la deficiencia de dopamina contribuye al deterioro intelectual. En la EP la reducción máxima de dopamina ocurre en la cabeza anterodorsal del caudado, que es el área que recibe proyecciones masivas de la corteza prefrontal y particularmente de la convexidad lateral. Lesiones experimentales en animales en la región anterodorsal del núcleo caudado causan dificultades en tareas que requieren de inhibición de la respuesta y formación y organización de planes. En contraste, las lesiones en la cola del caudado producen trastornos en la discriminación visual. Por lo tanto, aparentemente la concentración normal de dopamina dentro del estriado (especialmente dentro del núcleo caudado) asegura que los procesos cognoscitivos se mantengan intactos. La deficiencia de dopamina en el núcleo caudado afecta conductas que dependen del "circuito complejo" y los pacientes presentan una sintomatología frontal. La distribución de la dopamina residual dentro del estriado se vuelve crítica para las funciones cognoscitivas en términos de que afecta el circuito fronto-caudado dentro del "circuito complejo" que finalmente regresa la información procesada en el núcleo caudado a la corteza prefrontal. La sintomatología de tipo frontal se observa en una inhabilidad para ordenar y mantener programas cognoscitivos (actividades dirigidas hacia una meta) y por la presencia de signos motores de tipo frontal como la inhabilidad para mantener y organizar secuencias de acciones. En la exploración de estímulos visuales se observa segmentación y pérdida de la perspectiva figura-fondo.
También se ha encontrado que las células dopaminérgicas en el área ventral tegmental adyacente a la zona compacta de la substancia nigra, están involucradas en la patofisiología de la EP. Esta área da origen a la vía mesolímbicocortical que proyecta a áreas corticales (área frontal medial) y áreas límbicas (núcleo accumbens, amígdala, corteza cingulada, hipocampo, circunvolución paraolfatoria y séptum). Esta pérdida celular resulta en una reducción del 19% de la dopamina dentro de la convexidad lateral de la región prefrontal. En primates esta pérdida dopaminérgica produce trastornos en la inhibición y en programas de alternancia espacial.
A pesar de que la patología primaria de la EP es la degeneración de la proyección dopaminérgica al estriado, no todos los síntomas de estos pacientes se atribuyen a la pérdida de dopamina nigroestriatal. Existen otros sistemas neuroquímicos que se encuentran afectados como son células noradrenérgicas en el locus ceruleus; neuronas serotoninérgicas en el núcleo del rafé; acetilcolina por lesiones en el sistema septo-hipocampico y de la substancia innominada. A nivel cortical se ha reportado una reducción de somatostatina. La reducción de acetilcolina y de sus enzimas en el núcleo Basal de Meynert, ha sido asociada con los trastornos demenciales. En relación a los otros neurotransmisores, hasta la fecha, no se ha establecido una relación clara entre estos cambios bioquímicos y la sintomatología clínica, sin embargo, con base en datos que se han obtenido en la investigación con animales, se ha sugerido que la alteración selectiva de los sistemas noradrenérgicos podría causar trastornos en la atención; la reducción del metabolismo serotoninérgico se ha asociado con la depresión, y la disminución de somatostatina a nivel cortical se ha correlacionado con deterioro intelectual.
Aparentemente las lesiones en los diferentes sistemas neuronales no evolucionan en paralelo, pero son aditivas o se potencian una a otra en términos de expresión funcional. También, la variedad en extensión y grado de lesiones que se encuentran en EP podría ser el substrato patológico para la amplia variedad de síntomas motores y cognoscitivos que se observan.
DEMENCIA
El Manual de Diagnóstico y Estadística de los Desórdenes Mentales ( DSM-III-R ) , define que un paciente puede tener un diagnóstico de demencia si presenta, con un buen estado de alerta, una pérdida de sus funciones intelectuales y mnésicas y/o cambios de personalidad suficientemente severos que le impiden desempeñarse adecuadamente en su vida social y laboral. Además debe establecerse, la existencia de alguna etiología orgánica. Hasta hace poco, la demencia era considerada como un fenómeno unitario de un desorden cerebral pero, en estudios recientes se observa que la demencia presenta expresiones muy variables.
Neuropsicológicamente se han observado dos patrones de deterioro. El primero, es un patrón cortical que se expresa con deterioro intelectual, que incluye pérdida de habilidades en el lenguaje, amnesia, deterioro en sus habilidades visoespaciales, del aprendizaje y del cálculo, también se presenta agnosia y apraxia; las alteraciones en el área motora, sólo se observan en etapas finales de la enfermedad.
El segundo, es un patrón denominado subcortical que se caracteriza por trastornos en el habla, presentándose disartrias e hipofonía y lentitud para llevar a cabo las funciones mentales. Con respecto a la memoria sufren de olvidos y tienen dificultades para evocar información, existe un mal planeamiento de estrategias siendo difícil resolver problemas. También se observan alteraciones en habilidades visoespaciales y en tareas que requieren de atención. En cuanto a su estado de ánimo se ha observado falta de motivación, deterioro afectivo y desórdenes emocionales como apatía, depresión y como todos los desórdenes subcorticales presentan problemas extrapiramidales, caracterizados por movimientos involuntarios de tipo coreiformes, distonías, rigidez, temblor.
Se estima que la prevalencia de la demencia en la EP varía de acuerdo a la población estudiada, a la definición de demencia que se utilice y a las técnicas que se usen para la evaluación. Esto hace que las estimaciones de demencia en los pacientes con EP varíen entre 4% a 93 %.
Se ha reportado que la demencia en los pacientes con EP se caracteriza por: lentificación para iniciar actividades espontáneamente, incapacidad para resolver problemas, deterioro y disminución en la memoria, alteraciones en la percepción visoespacial, en la formación de conceptos, una pobre generación de palabras y una incapacidad para mantener el "set". Estos trastornos ocurren en la ausencia de afasia, apraxia, agnosia, desorientación, y/o indiferencia que típicamente se observa en la demencia cortical, como por ejemplo en la demencia senil de tipo Alzheimer.
Para documentar la existencia de demencia subcortical en la EP, se han llevado a cabo investigaciones sobre aspectos patológicos, bioquímicos y comportamentales, generalmente comparando pacientes con EP y con sospecha de DSTA, y apareados por edades con grupos controles de sujetos normales.
Algunos estudios han reportado que el perfil neuropsicológico de pacientes con EP es diferente del que presentan los pacientes con DSTA, y sugieren que existe una demencia subcortical que difiere de la demencia cortical. Como por ejemplo, en relación al lenguaje en los pacientes con EP se observan problemas de fluidez, pero no se han encontrado dificultades en la comprensión y en el uso apropiado del lenguaje. En contraste se ha encontrado que los pacientes con DSTA tienen dificultades con el uso y en la comprensión del lenguaje, pero no en la fluidez verbal. Otros estudios han reportado diferencias entre pacientes con EP con demencia y pacientes con DSTA en tareas de memoria inmediata, memoria remota, habilidades viso-espaciales, apraxia, fluidez, y depresión, y secuenciación. Sin embargo, otros estudios han encontrado que existen diferencias muy pequeñas entre los pacientes con EP demenciados y los pacientes con DSTA, por lo que han sugerido que pacientes con EP con demencia no presentan una demencia subcortical única sino que lo que presentan es un subtipo de EP con una coexistente DSTA.
En general estos estudios indican que existe un subgrupo de pacientes de EP con demencia, la cual puede o no coexistir con DSTA concomitante. La única evidencia que existe para poder diferenciar subtipos de EP, es que los pacientes con esta enfermedad que presentan deterioro cognoscitivo global son más viejos, tienen un inicio de síntomas tardíos muestran evidencia tanto de demencia subcortical y atrofia frontal cortical y no responden tan efectivamente a la L-Dopa y al deprenil como los pacientes con EP que no presentan demencia. Sin embargo otros estudios no han encontrado diferencias de edad entre pacientes dementes y no dementes. Pirozzolo y Colbs., aportaron un gradiente de déficits cognoscitivos sin ninguna evidencia de un subtipo con demencia. Huber y Colbs., diseñaron una batería para diferenciar entre demencia subcortical y demencia cortical, y encontraron diferencias significativas entre los grupos. Los pacientes con EP mostraron puntajes más altos que los pacientes con DSTA en varias pruebas de la batería.
En relación a los estudios patológicos podemos decir que la evidencia experimental que apoya el argumento de que los pacientes con EP y demencia tienen una DSTA concomitante se basa en estudios que han documentado que la patología de DSTA se presenta con más frecuencia en pacientes con EP que en sujetos normales de las mismas edades. Varias investigaciones han reportado que los pacientes que presentan el criterio histológico de la EP (cuerpos de Lewy en la sustancia nigra o en locus coeruleus), muestran cambios patológicos característicos de la DSTA, especialmente placas seniles en el hipocampo. En contraste, De la Monte y colbs., demostraron diferencias patológicas distintivas entre pacientes clasificados con EP y demencia y los clasificados con EP y DSTA. Esto es, los pacientes con EP y DSTA mostraron la misma patología subcortical que la de los otros tipo de pacientes, pero además mostraron atrofia global en la corteza cerebral y en la sustancia blanca, y una gliosis significativa a través de toda la corteza . Sin embargo estos estudios no describieron los criterios utilizados para diagnosticar demencia, ni como fueron agrupados los sujetos en grupos de EP con demencia vs. EP con DSTA. Por lo tanto estos estudios únicamente pueden ser interpretados como una evidencia de la existencia de un subtipo de pacientes con EP que pueden tener DSTA concomitante.
Otros autores plantean que muchos pacientes con EP con demencia no presentan cambios patológicos del tipo de demencia de Alzheimer. Heilg y colbs., describen 5 casos de pacientes con EP demenciados con alteraciones mínimas en la corteza o en el hipocampo y con severa patología en la substancia negra y el locus coeruleus. El análisis neuropatológico incluyó regiones frontales, pariretales, temporales, occipitales, hipocampo, ganglios basales, cerebro medio, puente y médula espinal . Los resultados no mostraron cambios patlógicos caracteristicos de la DSTA, por lo los autores atribuyen la demencia a la patología subcortical.
Otro método para investigar la patología responsable de los síntomas cognoscitivos de la EP es la utilización de tomografía por emisión de positrones (PET). Estos estudios han revelado que en los pacientes con EP existe una reducción en la formación de dopamina en el putamen, indicando deficiencias en el almacenamiento y formación de dopamina subcortical. Es de gran interés el hecho de que en los pacientes con EP se ha observado una disminución metabólica en corteza frontal, sugiriendo así el involucramiento córtico-frontal en los desórdenes cognoscitivos (44).
Algunos autores han sugerido que los cambios cognoscitivos no son debido a una anormalidad cortical sino que son secundarios a una disfunción de los ganglios basales y del sistema dopaminérgico. Por ejemplo Mortimer y col( 5) y Martilla y Rine (45) apuntaron una asociación entre la existencia de demencia y el grado de rigidez y bradiquinesia sugiriendo la posibilidad de una etiología común entre síntomas motores y cognoscitivos. Sin embargo, Portin y Rinne (36), en un estudio longitudinal en el que se hizo un seguimiento de pacientes con EP durante 8 -10 años, concluyeron que las funciones motoras y cognoscitivas en la EP no se correlacionan. En su estudio encontraron que el tratamiento con L-dopa afecta a los síntomas cognoscitivos en forma transitoria y en menor grado que a los síntomas motores. Evidencias recientes reportan que la medicación dopaminérgica no afecta a los trastornos demenciales y que en los pacientes con EP demenciados se observa una reducción en la actividad colinérgica, lo que sugiere que existe un subgrupo de pacientes con EP demenciados y que estos pacientes, así como los pacientes con DSTA presentan un decremento en la actividad colinérgica ( 35).
Como mencionan Raskin y Colbs., (46) los estudios patológicos y bioquímicos que se han realizado en pacientes con EP, muy pocas veces incluyen observaciones comportamentales, por lo tanto se dificulta llegar a alguna conclusión en cuanto a las relaciones existentes entre aspectos físicos y cognoscitivos de la enfermedad.
Los estudios que han evaluado las alteraciones cognoscitivas no han realizado estudios patológicos ni un seguimiento a largo plazo, por lo que las únicas conclusiones que a la fecha se pueden hacer son:
1. Algunos pacientes demenciados presentan cambios cerebrales parecidos a los que presentan los pacientes con DSTA.
2. Los pacientes con demencia parecen constituir sólo un subtipo de demencia, pero la etiología de la demencia aún no es clara.
TRASTORNOS COGNOCITIVOS ESPECIFICOS
Aunque algunos pacientes con EP presentan cambios drásticos a nivel intelectual, en otros sólo se observa la presencia de una sintomatología cognitiva específica que incluye: sintomatología de tipo frontal, fallas en la memoria, en el procesamiento visoespacial, en el lenguaje y en el habla y depresión.
Funciones Viso-Espaciales
Existen diversos estudios que sugieren la presencia de déficits visoespaciales en pacientes con E.P.. Algunos investigadores afirman que posiblemente la función más afectada en pacientes con EP se refiere a los procesos visoespaciales. Sin embargo, en este tipo de evaluaciones es importante aclarar que la exploración de funciones visoespaciales es muy heterogénea ya que involucra diversos aspectos como exploración espacial, discriminación visual, orientación espacial, percepción de ángulos, memoria topográfica, percepción espacial, y habilidades construccionales entre otras.
Bentin, Silverberg y Gordon estudiaron 32 pacientes con E.P. y encontraron dificultades en la orientación, reconocimiento de caras, patrones visuales y dibujos o escenas temáticas. Estas deficiencias habían sido reportadas por Teuber y Proctor en monos con lesiones en el putamen y en el hombre con lesiones frontales. Estos autores señalan que la alteración básica se debe a una dificultad en la orientación espacial y visopostural las cuales dependen de las conexiones entre los ganglios basales y la corteza frontal, sugiriendo que el patrón de deficiencias se parecen a las alteraciones que presentan pacientes con lesiones en el hemisferio derecho.
Diversos estudios han demostrado que los pacientes con EP presentan un déficit viso-espacial aún en tareas en donde se requiere de una respuesta manual y/o motora mínima. La ejecución de los pacientes con EP, fue significativamente peor que la de los controles apareados por edad en las pruebas de matrices progresivas del Raven (38), que es una prueba de razonamiento y en la prueba de discriminación visual de formas de Benton (39,5)
Bowen y colbs., apuntaron un estudio en el cual los sujetos tenían que colocar en una representación ventral y dorsal de la figura humana diferentes partes del cuerpo. Los pacientes con E.P. tuvieron más errores en la confusión derecha-izquierda, especialmente en la figura que requirió que el sujeto invirtiera su propia imagen corporal.
Taylor, Saint-Cyr y Lang utilizando pruebas de figuras superpuestas, de razonamiento numérico en relaciones espaciales, discriminación figura-fondo, rotación mental y orientación izquierda-derecha. Compararon 40 pacientes con E.P. con 10 sujetos controles y los resultados no mostraron diferencias significativas entre los dos grupos pero, al comparar el tiempo de ejecución se encontraron diferencias en la gran mayoría de las pruebas, excepto en orientación derecha-izquierda; también se observaron diferencias entre hombres y mujeres con E.P., encontrando que los hombres realizaron en menor tiempo tareas como razonamiento numérico, relaciones espaciales, discriminación figura-fondo y orientación izquierda-derecha; mientras que las mujeres lo hicieron sólo en tareas de rotación mental.
Hovestadt, Jong y Meerwaldt estudiaron 44 pacientes con E.P. a quienes se les aplicaron cuatro pruebas que evalúan orientación espacial: La prueba de orientación (ROT), la prueba de orientación de líneas, la prueba de reconocimiento facial y la Escala de Inteligencia para Adultos Weschler (WAIS). Los resultados mostraron que 43 pacientes tuvieron un deterioro en la prueba de orientación, 7 en la prueba de orientación de lineas y 17 fallaron en la prueba de renocimiento facial. Estos resultados no se correlacionaron con la edad, la duración de la enfermedad, los puntajes del WAIS ni con la severidad del deterioro motor.
En relación a la orientación personal y extrapersonal se ha llegado a la conclusión de que los pacientes con EP tienen un déficit específico en orientación espacial y tienen dificultades para manejar cambios espaciales. Diversos investigadores han descubierto que los pacientes con EP tienen problemas para juzgar la orientación vertical tanto en forma visual como postural. Ransmayr y colbs., (1987) apuntaron que la dificultad en el apareamiento de ángulos en orientación vertical es uno de los primeros síntomas de daño cognoscitivo en la EP. Bowen y colbs., realizaron una prueba que consistió en que los pacientes tocaran las partes de su cuerpo que se iban señalando en un diagrama. En general, los pacientes con síntomas predominantes de lado izquierdo y bilaterales cometían más errores que los pacientes con síntomas de lado derecho. Sin embargo, estos errores sólo sucedían cuando el cuerpo en el diagrama se veía de frente.
Después de varios estudios en este campo se concluyó que:
1. Los pacientes con EP con síntomas predominantes en el lado izquierdo pueden diferir comportamentalmente de aquéllos con síntomas predominantes en el lado derecho.
2. Que los pacientes pueden tener déficits específicos en cuanto a mantener la orientación espacial, particularmente cuando se requiere que se haga un cambio en la orientación.
Los resultados de estos estudios sugieren que los pacientes con EP tienen un déficit viso-espacial tanto en tareas que requieren respuestas motoras como las que no. Los déficits más claramente observados involucran tareas de orientación espacial y corporal pero no de rotación mental. Aún no está claro si existe un déficit específico en orientación espacial mediado por los ganglios basales, o como mencionan Brown y Marsden y otros autores se debe a un déficit para cambiar de set mental, que, tal vez refleja una disminución en la estimulación de los ganglios basales a la corteza frontal. Este interrogante no puede ser fácilmente aclarada ya que muchos de los estudios utilizan tareas que miden varias funciones y no describen cuidadosamente la población en términos de variables como severidad de la enfermedad y síntomas motores del lado predominante.
Sintomatologia de tipo frontal
Muchas investigaciones han observado íntima conexión de los ganglios basales a la parte inferior de los lóbulos frontales y el decremento de salidas dopaminérgicas a estas regiones en casos de pacientes con Parkinson. Estas investigaciones han sugerido que los déficits cognoscitivos en la EP no reflejan problemas en el funcionamiento de los ganglios basales sino una desconexión a los lóbulos frontales. Se ha sugerido que las deconexiones fronto-talámicas explican la pérdida de espontaneidad e imaginación y la falta de iniciativa que presentan los pacientes con EP. En un cuestionario de funciones cotidianas los pacientes con EP fueron clasificados diferentes a los normales en conductas relacionadas a la iniciativa, conductas estereotipadas, indiferencia, desinterés, dependencia social, y control intelectual. El enlentecimiento para dar respuestas y la tendencia a verbalizar pero no ejecutar movimientos correctos puede también dar evidencia del involucramiento de los lóbulos frontales en la EP. Existen también otras evidencias empíricas que señalan deficiencias en funciones mediadas por los lóbulos frontales, como son las respuestas demoradas y cambios en el set mental.
Evidencias experimentales con monos en los que se utilizaron tareas de respuestas demoradas sugieren que las lesiones en los ganglios basales producen síntomas muy parecidos a los que se observan después de la ablación bifrontal cortical. La estimulación eléctrica del caudado también ocasiona alteraciones en la ejecución de respuestas demoradas en monos. Los pacientes con EP también han demostrado dificultad en este tipo de tareas, aunque ésto puede presentarse en aquéllos pacientes con déficits cognoscitivos severos. En estas tareas de respuesta demorada se observó que aquéllos pacientes con EP que habían padecido talamotomia tenían más dificultades en tareas de apareamiento visual que los sujetos normales. Los déficits en estas clases de tareas están asociados con patología en las proyecciones principales de los núcleos dorsomediales del tálamo a los sistemas frontales dorsolaterales y orbitales.
Existen varios estudios que apoyan la teoría de que los trastornos cognoscitivos en la EP se deben a la desconexión de las vías frontales. Taylor, Saint-Cyr y Lang utlizando el Wisconsin Card Sorting Test (WCST), compararon 40 pacientes con EP y 40 sujetos normales, encontraron diferencias significativas entre los números de categorías, caracterizándose la ejecución de los pacientes con EP por un predominio de respuestas perseverativas, así como un mayor número de errores para llegar a la primera categroria, que implica una menor habilidad para elaborar un plan de acción ante una tarea. Flower y Robertson (57)diseñaron la prueba "Odd-Man-Out" como una medida de mantenimiento del set mental que puede ser más sensible que el WCST. Esta prueba requiere que el sujeto indique cual del conjunto de letras o números es diferente de otros conjuntos basados en una o dos reglas posibles , los sujetos tienen que utilizar las dos reglas alternativas en ensayos sucesivos. Los pacientes con EP ejecutan solo ligeramente más deficiente que los controles normales, lo que indica una buena habilidad para la ejecución de esta tarea, sin embargo sus patrones de errores fueron diferentes de los controles. Los pacientes con EP cometían más errores durante todos los ensayos mientras que los controles sólo cometían errores en los ensayos iniciales. Los pacientes cometían errores en ensayos que inicialmente habían realizado correctamente lo que indica fluctuaciones en la ejecución y una tendencia a regresar a las reglas precedentes.
Se ha observado que en pruebas de fluidez verbal los pacientes con EP producen un número mayor de palabras que los pacientes con daño en los lóbulos frontales. Se ha encontrado, además que los pacientes con EP tienen un rango normal de fluidez verbal en pruebas que requieren la generación de palabras que comienzan con una letra en particular . Sin embargo la ejecución en tareas de fluidez que requieren de la generación de palabras dentro de una categoría semántica específica está afectada.
Se han encontrado dificultades similares en tareas que requieren de dos movimientos motores simultáneos. Estos déficits han sido interpretados como reflejo de daño en lóbulos frontales. Taylor y colbs., sugieren que el área motora suplementaria de lóbulos frontales tiene una función especializada en el planeamiento de éstos movimientos.
Memoria y Atención
Diversos estudios han reportado alteraciones en la memoria en los pacientes con E.P., aún cuando, existe controversia respecto a esto, sobre todo debido a la pruebas utilizadas, a la población estudiada ó también diversas hipótesis explicando las razones por las cuales lo pacientes presentan alteraciones en la memoria.
Algunos autores han estudiado diferentes procesos específicos de los trastornos de memoria. Por ejemplo Bowen y Colbs., aportaron un déficit para evocar inmediatamente un pasaje lógico pero, la memoria a largo plazo se encontró intacta.
Taylor y Colbs., en un estudio con 40 pacientes con E.P. aplicaron la Escala de Memoria de Weschler y encontraron diferencias significativas en memoria lógica, en la reproducción inmediata, pero no en la evocación diferida. Estos autores proponen que los parkinsonianos pueden inicialmente codificar información adecuadamente, pero la organización y la consolidación requieren de más tiempo para que se activen estrategias de búsqueda, por lo que los pacientes, aparentemente presentan una ejecución adecuada en tareas de memoria, particularmente si tienen el tiempo suficiente para consolidar la información.
Con la evaluación de memoria inmediata utilizando Memoria de dígitos de la Escala de Memoria de Weschler en pacientes con E.P. se han reportado datos discrepantes. Algunos autores refieren un deterioro en la ejecución de memoria mientras que otros reportes indican una ejecución normal en este tipo de tareas.
En lo que respecta a la memoria remota, Sagar y colbs., descubrieron un deterioro selectivo al recordar datos de información sobre alguna escena. Aunque, Freedman y colbs., no encontraron fallos en tareas que evalúan memoria remota utilizando la prueba de "caras famosas" en pacientes sin demencia; no así en los pacientes con demencia en los que se observó un deterioro significativo muy similar al reportado en pacientes con demencia de tipo Alzheimer.
Huber, Shuttleworth y Paulson aplicaron la prueba de atención de Albert, la prueba de retención de dígitos y la de pares asociados a 31 pacientes con E.P. (20 con demencia y 11 sin demencia) y a 18 sujetos controles. No encontraron diferencias significativas en la memoria inmediata, pero la memoria a corto plazo se vio decrementada en los pacientes con E.P., mientras que la memoria remota estuvo afectada en diferentes grados en los pacientes con E.P demenciados.
En cuanto a la memoria visual aparentemente no se han apuntado alteraciones. Flowers, Pearce y Pearce estudiaron a 54 pacientes con E.P. y los compararon con un grupo control; en este estudio no encontraron diferencias significativas en prueba de reconocimiento visual inmediato y demorado, sin embargo, los pacientes con E.P. obtuvieron puntuaciones más bajas que los controles.
Con los datos anteriores, autores como Brown y Marsden propusieron un patrón patológico de la memoria en la E.P., en el que sugieren que los pacientes presentan fallos principalmente en la memoria activa que requiere que el sujeto manipule el material. En cambio, en tareas que evalúa la memoria pasiva en la cual se necesita que el sujeto manipule el material mentalmente, no se observan diferencias. Estos datos implicarían que la alteración no se encuentra en la recepción de la información sino en la consolidación de ésta.
Las tareas en las que se implementa un retardo después del reconocimiento pueden requerir un cambio entre diferentes almacenamientos. Si el retardo es de corta duración, alguna información puede aún estar en uno de los almacenamientos de memoria a corto-plazo, mientras otras partes de información han sido codificadas en los almacenamientos de más largo plazo, estos reconocimientos por lo tanto, requieren la integración de ambos almacenamientos. Cuando el período de retardo es suficientemente largo, toda la información se almacena a largo plazo y no es necesaria la integración. Se han reportado diferencias entre los pacientes con EP y los controles normales en tareas que requieren evocar. De Lancey Horn encontró que los pacientes con EP tenían ejecuciones buenas al igual que los controles en tareas de evocación que involucraban caracteres chinos, pero la naturaleza de su ejecución era diferente a la de los controles. Los sujetos controles reconocían más rápidamente cuando un reactivo de la prueba estaba precedido por un retardo que cuando no era así. En los pacientes con EP no se observó esta diferencia.
Estos hallazgos sustentan la hipótesis que los pacientes con EP presentan déficit de memoria sólo cuando la tarea requiere integración entre almacenamientos de memoria. Tweedy, Langer y McDowell encontraron déficit cuando se presenta un intervalo antes del reconocimiento. En contraste, Flowers y Robertson no encontraron diferencias entre controles y pacientes con EP en tareas con un intervalo de 45 minutos, suficientemente largo para transferir toda la información al almacenamiento de largo plazo. Sullivan y Sagar encontraron que los pacientes con EP, que tenían problemas en reconocimiento inmediato, mejoraban su ejecución con un intervalo sustentando así la idea de que la ejecución mejoraba una vez que toda la información había sido transferida a un sistema de almacenamiento.
En general, los pacientes con EP ejecutan dentro de los límites normales en las pruebas que involucran procesos automáticos, reconocimiento inmediato, acceso a la memoria semántica a largo plazo, rastreo de memoria a corto plazo. Sin embargo, los pacientes con EP han demostrado dificultades en tareas que requieren reconocimiento tardío, evocación inmediata o tardía. Estos resultados apoyan la idea que los déficits de memoria en EP son aparentes únicamente cuando la tarea requiere de la integración de información de diferentes sistemas de almacenamiento de memoria y no cuando se requiere del almacenamiento y evocación de información per se.
Lenguaje y habla
En estudios neuropsicológicos casi no se reportan problemas de lenguaje en pacientes con E.P., aparentemente se encuentran intactos en aspectos lógico-gramaticales y lógico-verbales complejos, pero tienen dificultades para realizar análisis detallados ó estrategias adecuadas para solucionar y verificar problemas. La enfermedad involucra básicamente el componente motor tanto en sus aspectos articulatorios, la velocidad y el aspecto entonacional como el volumen del lenguaje. No se observan alteraciones en los diferentes niveles integrativos del lenguaje, por lo que las alteraciones motoras, incluyendo la reducción y la lentificación están vinculadas al sistema de ejecución a nivel subcortical.
Trastornos afectivos y depresión
Se han observado cambios diversos en la personalidad del paciente con E.P. ya que conforme va progresando la enfermedad sufren una pérdida de interés por la familia, por sus amigos, disminuyen su convivencia y evitan tener nuevas relaciones interpersonales. Algunos autores refieren una disminución evidente y dramática en la actividad social y se ha postulado que los pacientes sufren un envejecimiento social prematuro.
Existen estudios que se han realizado con el fin de conocer el perfil de personalidad en pacientes con E.P. Hoehn, Crowley y Rutledge aplicaron el Inventario Multifásico de la Personalidad (MMPI) y encontraron una elevación en las escalas de depresión, hipocondriasis y esquizofrenia, que se caracterizó con una tendencia a ser sujetos tensos, preocupados, con normas sociales rígidas y con tendencia a disminuir sus expresiones emocionales.
La apatía, la inercia y una gran variedad de conductas y cambios de la personalidad han sido descritos como síntomas dentro de los desórdenes subcorticales. En estos cambios conductuales, la depresión ha sido considerada como una alteración no cognitiva en los desórdenes subcorticales.
La asociación entre E.P. y depresión ha sido reportada frecuentemente ;no obstante su prevalencia exacta, su naturaleza y etiología es desconocida, así como su relación con el tiempo de evolución de la enfermedad, con el inicio de ésta y con otras alteraciones cognoscitivas con las que se acompaña.
Las estimaciones de depresión en la EP varia del 12% al 90%. En una revisión de 14 estudios, Gotham,Borown y Marsden apuntaron una media estimada de 46%. Las razones de este amplio rango, incluye diferencias demográficas de los pacientes (ej: pacientes hospitalizados y externos) y la presencia de muchos cambios físicos comunes en la EP que aparecen como pruebas somáticas complementarias de las escalas de depresión como por ejemplo: problemas en la postura, enlentecimiento motor, reducción de la expresión facial. Por ello estos autores sugieren que el diagnóstico psiquiátrico de la depresión en muchos estudios se basa no solamente en cambios en el estado de ánimo, sino también en características físicas y conductuales tales como disturbios en la postura, en la actividad motora, en la expresión facial, en el tono de la voz, alteraciones en el patrón de sueño, en el apetito y en la libido, pero muchas de esas características son comunes en la E.P. debido a los trastornos motores, sin que halla datos que sugieran la presencia de depresión.
Existen hipótesis que postulan una relación entre el deterioro cognitivo y la depresión, Starkstein y colbs., estudiaron 105 pacientes con E.P. a los cuales les realizaron evaluaciones neurológicas, psiquiátricas y neuropsicológicas, encontrando que la severidad de la depresión es uno de los factores más asociados con la grado de deterioro cognitivo. Los pacientes con depresión mayor tuvieron una mala ejecución en la pruebas neuropsicológicas comparados con los pacientes no deprimidos.
Taylor,Saint-Cyr y Lang para explicar lo anterior compararon a 30 pacientes con E.P. (divididos en dos grupos, 15 pacientes con depresión y 15 sin depresión), a 15 pacientes psiquiátricos con depresión endógena y a 15 sujetos controles. A todos ellos se les aplicó la Escala de Memoria de Weschler y algunas pruebas del WAIS-R. Los resultados indicaron que los pacientes con E.P. tuvieron una ejecución mejor en la pruebas tanto de memoria como en el WAIS-R que los sujetos controles y ambos grupos obtuvieron puntajes mejores que los pacientes con depresión endógena. Estos autores concluyen que la labilidad emocional que presentan los pacientes con E.P. forma parte de un proceso patofisiológico el cual involucra áreas corticales prefrontales.
Otros autores se han abocado a contestar una pregunta ¿La depresión en la E.P. es una depresión reactiva o una depresión endógena? considerando que la misma enfermedad puede ser la causa de la depresión, ya que es una enfermedad crónica, progresiva e incapacitante.
Se ha encontrado una alta incidencia de depresión en pacientes con enfermedades crónicas. Para averiguar si la depresión en la E.P. es producto de un estado crónico, Gotham, Brown y Marsden estudiaron a tres grupos de pacientes: 189 pacientes con E.P., 121 pacientes con diagnóstico de artritis y 100 sujetos controles. A todos los sujetos les aplicaron cuatro pruebas, el Inventario de Depresión de Beck, La Escala de Hopelessness Beck, el Indice de Ansiedad de Spielberger y un Cuestionario de actividades de la vida cotidiana. Encontraron que los pacientes con E.P. estaban más deprimidos que los controles; y el nivel de depresión no fue tan alto al compararlo con pacientes con enfermedades crónicas.
Starkstein y Colbs. llevaron a cabo un estudio con 105 pacientes con E.P., 21% fueron diagnosticados con depresión mayor, 20% con un diagnóstico de depresión menor y el resto no presentó depresión. Los resultados obtenidos indicaron que factores como historia familiar de desorden psiquiátrico, funcionalidad social, severidad del temblor, rigidez y aquinesia no mostraron asociación significativa con la depresión; no obstante, los pacientes deprimidos tuvieron una correlación significativa entre deterioro en actividades cotidianas y funciones cognitivas, en comparación con los pacientes no deprimidos.
Bieliavskas y Glantz estudiaron 33 pacientes con E.P. a los que evaluaron neurológica y neuropsicológicamente. En el 70% de los pacientes, la depresión no estuvo relacionada con la presencia de demencia ni con la duración de la enfermedad ni con la edad ó con índices relacionados con daño frontal. Estos datos apoyan la hipótesis que sugieren que la depresión es reactiva a la enfermedad. Sin embargo, otros estudios no han encontrado asociación entre severidad de los síntomas ni con respuesta al médicamente con la depresión.
La E.P. ha sido asociada con disturbios neuroquímicos a nivel de neurotransmisores como la serotonina, la cual ha sido involucrada en modelos de depresión. Se ha postulado que la depresión refleja cambios bioquímicos y neuroanatómicos que son intrínsecos a la EP. y se ha propuesto que existe un subgrupo de pacientes con EP que presentan un decremento en los niveles de serotonina así como de dopamina, y que esta deficiencia de serotonina es la que conlleva a los estados depresivos. Esto ha sido reforzado por el descubrimiento de bajas concentraciones de ácido 5-hidroxindoleacético en el fluido cerebro espinal de pacientes con EP diagnosticados como depresivos. Como sugieren Gotham, Brown y Marsden, la depresión en la E.P puede deberse a diversas etilogias como son : alteraciones en los niveles cerebrales de aminas (algunos autores consideran a la depresión como una parte integral de un desorden en los ganglios basales), como una reacción a una enfermedad incapacitante, a ambos ó a otros factores no relacionados con la enfermedad .
Todos los datos hacen pensar que la E.P. es una enfermedad progresiva y degenerativa que involucra un cuadro característico y sugiere que la E.P. no es una enfermedad única sino que es todo un complejo en el que pueden estar interviniendo diversos sistemas neurofisiológicos, bioquímicos y patológicos que interactúan como un todo.
En conclusión podemos decir que los trastornos cognoscitivos forman parte de la sintomatología clínica de la EP. Estos trastornos pueden presentarse en grado variable. En algunos pacientes se han encontrado trastornos cognoscitivos específicos mientras que en otros se ha notado la presencia de un deterioro más generalizado reportándose un cuadro demencial . La demencia o el deterioro irreversible de las funciones intelectuales, incluyendo memoria, cognición y percepción, se observa en aproximadamente 30% de los pacientes con EP. La depresión ha sido frecuentemente asociada con la EP, sin embargo, los reportes acerca de su incidencia han sido controvertidos y varían entre un 20% y 60% . A pesar de que los efectos de la enfermedad pueden contribuir a los patrones depresivos, se han utilizado otros factores neurofisiológicos para explicar la depresión. Se ha sugerido una degeneración de las vías dopaminérgicas mesolímbicas y mesocorticales y han reportado un aumento de serotonina en pacientes con EP que presentan depresión.
Las bases patofisiologicas de los trastornos cognoscitivos en la E.P. es controversial y aún inexacta. Diversas líneas de evidencia sugieren que las deficiencias dopaminérgicas contribuyen al deterioro intelectual, sin embargo en algunos casos se ha encontrado que la perdida dopaminérgica se acompaña de deficiencias colinérgicas y en otros neurotransmisores y esta combinación de anormalidades neuroquímicas exagera los trastornos intelectuales
La literatura concerniente a la neuropsicología de la EP sugiere que mientras ciertos cambios cognoscitivos son frecuentemente observados en pacientes con EP, otros cambios cognoscitivos son únicamente observados en subgrupos particulares de pacientes con EP. La relación entre los factores causales de la enfermedad, así como la neuropatología, las variables individuales, y la presencia de estos subtipos requieren más investigación.
Los síntomas neuropsicológicos más comúnmente reportados en un gran número de pacientes son déficits en orientación espacial, dificultad en el cambio de un set mental, déficits en memoria, reducida fluidez verbal y dificultades en la iniciación. Se sugiere que las dificultades para cambiar el set mental, las dificultades en las respuestas demoradas y la reducida fluidez, son parte de un síndrome simple que refleja falta de conexiones a la corteza cerebral frontal. La sintomatología de tipo frontal se observa en una inhabilidad para ordenar y mantener programas cognoscitivos (actividades dirigidas hacia una meta) y por la presencia de signos motores de tipo frontal como la inhabilidad para mantener y organizar secuencias de acciones. En la exploración de estímulos visuales se observa segmentación y pérdida de la perspectiva figura-fondo .
Además de estos síntomas comúnmente reportados, se han observado otros déficits en subgrupos particulares de pacientes con EP. Existe evidencia de un subgrupo de pacientes con EP con demencia, y se sugiere que este tipo de pacientes tienen una DSTA concomitante. Existen además otras evidencias que sugieren que existe un subgrupo de pacientes que presentan depresión pero ésta no es sólo una respuesta a la enfermedad.
Los estudios revisados indican claramente la necesidad de llevar a cabo una evaluación neuropsicológica en los pacientes con EP para determinar los síntomas particulares.
3.1.3- TRATAMIENTO
El tratamiento médico de la enfermedad de Parkinson se realiza para controlar los síntomas, supliendo la alteración de los transmisores.
Se suele usar Levodopa , que es la molécula que el cerebro utiliza para producir Dopamina, con ello se mejora la coordinación de movimientos, se suele asociar con otros medicamentos agonistas dopaminérgicos (Carbidopa).
La amantadina se utiliza para tratar el temblor.
Deben tratarse también otros problemas médicos generales por lo que debe existir una buena relación con el médico de cabecera. A veces se asocian antihistamínicos, antidepresivos, bromocriptina, IMAO, y otros medicamentos para tratar otros síntomas.
En cada caso el tratamiento farmacológico será individualizado, según las características del paciente y los síntomas predominantes en cada caso.
La realización de un programa de ejercicios físicos es muy recomendable, así como el apoyo y tratamiento psicológico de la situación.
Existen en marcha ciertos procedimientos quirúrgicos que modifican las alteraciones cerebrales, si el caso es muy intenso y no mejora con tratamiento, ésta posibilidad debe de ser consultada con su especialista.
3.1.4- PRONÓSTICO
El Parkinson no tratado es incapacitante y lleva a una muerte prematura. Los pacientes tratados mejoran claramente de los síntomas, la variabilidad de respuesta a los tratamientos es diversa y por ello el pronóstico de cada caso dependerá de la tolerancia a los tratamientos y su eficacia en cada caso.
Los problemas del tratamiento son:
-
Movimientos involuntarios
-
Nauseas o vómitos
-
Sequedad de mucosas
-
Cambios en comportamiento
-
Desorientación o confusión mental
-
Alucinaciones
-
Perdida de funciones mentales
En la enfermedad de Parkinson y en los “parkinsonismos” se lesionan determinadas zonas del sistema nervioso que normalmente controlan y coordinan los movimientos.
La zona que más se altera es la sustancia nigra , un pequeño núcleo situado en el mesencéfalo, relleno de neuronas más oscuras de lo normal. Al lesionarse la sustancia nigra, van muriendo sus neuronas que producen dopamina. La dopamina es una sustancia química que utilizan algunas neuronas para conectarse entre sí. En personas normales, las neuronas de la sustancia nigra emiten unas largas prolongaciones (axones) para conectarse con los núcleos estriados, y lo hacen utilizando la dopamina.
El tratamiento médico de la enfermedad de Parkinson sigue siendo sintomático pero ha progresado notablemente en los últimos años. En la mayor parte de los casos evolucionados debe combinarse levodopa con agonistas dopaminérgicos además de otros fármacos para los diversos problemas asociados que pueden presentarse. En determinados casos hay indicaciones quirúrgicas. Deben tratarse también otros problemas médicos generales por lo que debe existir una buena relación con el médico de cabecera.
Es imprescindible que el tratamiento farmacológico sea individualizado, según las características del paciente y las peculiaridades de su situación. Es muy importante la rehabilitación física, la atención psicológica y las medidas de inserción social. Las revisiones periódicas deben atender no sólo a los síntomas motores sino a los numerosos problemas concretos que pueden parecer leves pero que resultan muy molestos para el paciente (exceso de saliva, estreñimiento, caídas de tensión arterial, depresión, trastornos del sueño, etc.).
Los nuevos sistemas de comunicación permiten mejorar la atención médica abriendo nuevas vías y posibilidades.
En el caso concreto de la enfermedad de Parkinson muchas de las consultas se limitan a pequeñas modificaciones de dosis o a la resolución de un problema concreto (por lo general leve pero molesto) que ha aparecido. Si el médico conoce bien al paciente puede ser suficiente con una llamada telefónica o una comunicación escrita.
El tratamiento convencional de la enfermedad de Parkinson (EP) consiste en la utilización de Levodopa y otros fármacos, que intentan compensar el déficit dopaminérgico responsable de la mayor parte de los síntomas de la enfermedad. Actualmente el tratamiento farmacológico no permite compensar la enfermedad indefinidamente. En al menos el 50% de los pacientes tratados con fármacos surgen importantes complicaciones en forma de fluctuaciones de la movilidad, movimientos involuntarios y alteraciones psiquiátricas, que perturban gravemente el control terapéutico y conllevan una importante incapacidad funcional.
Adicionalmente, el cuadro clínico se agrava con la existencia de síntomas con menor o escasa respuesta farmacológica, tales como los trastornos de la marcha, el lenguaje y el equilibrio.
El desarrollo de los fármacos antiparkinsonianos en los últimos 25 años ha intentado modificar estas complicaiones motoras y ofrecer una reducción satisfactoria de las manifestaciones cardinales de la enfermedad, generando combinaciones ingeniosas de levodopa y sintetizando nuevas moléculas que mimeticen la actividad de la levodopa, aplicándolos por diferentes vías y métodos, logranbdo el control parcial y temporal de esas complicaciones y con frecuentes efectos colaterales intolerables.
Hasta el momento han sido ensayados o están en ensayo mas de 100 compuestos farmacológicos y la regla general es que los productos que mejoran los síntomas cardinales inducen o se asocian a complicaciones. Desde su introducción hace 30 años la levodopa se mantiene como la principal estrategia terapeútica farmacológica en el tratamiento de la EP, por su demostrada eficacia en el control sintomático, su influencia en el incremento de la calidad de vida de estos pacientes y por su demostrada capacidad de incrementar la expectativa de vida de los mismos. En consecuencia, los esfuerzos dirigidos a disminuir las complicaciones relacionadas a su uso crónico, han generado una variedad de compuestos dirigidos a estabilizar su concentración en sangre y a incrementar su permanencia y efecto en la sinapsis. Sin embargo, aunque con resultados parciales, no se atisba ningún producto o estrategia de combinación que resuelva la limitación originada por los efectos del uso crónico de levodopa. Por el contrario, con la progresión de la enfermedad aumentan las demandas del precursor de dopamina y con ello su toxicidad.
En resumen existen dos tendencias farmacológicas bien establecidas:
Disminuir la velocidad de progresión de la enfermedad.
Control sintomático incluida la eliminación de las complicaciones asociadas al tratamiento farmacológico crónico.
La imposibilidad de lograr esos objetivos en la actualidad por métodos farmacológicos ha generado la tendencia a evaluar la cirugía como tratamiento alternativo; las razones fundamentales de este renovado interes por la cirugía estereotáxica pueden resumirse en los siguientes apartados.
Falta de prespectivas farmacológicas a medio plazo que permitan tratar o prevenir las complicaciones asociadas con la levodopoterapia crónica.
Un mayor conocimiento fisiopatológico de las estructuras implicadas en el origen del síndrome parkinsoniano.
Notables avances técnicos que permiten definir la estructura deseada y su relación especial con estructuras vecinas, así como su nivel de actividad.
La cirugía funcional estereotáxica como opción terapeútica en la EP se remonta a 1939. Los conocimientos generados a partir del estudio de los ganglios basales y el desarrollo de la exploración fisiológica de la actividad neuronal en animales de experimentación y en enfermos parkinsonianos han permitido establecer una teoría más coherente sobre la disfunción de los circuitos motores en esta enfermedad. Estos elementos ofrecen un sustento fisiopatológico más racional para el abordaje quirúrgico focal.
Por su parte, el desarrollo tecnológico ha condicionado una mayor resolución en la visualización de esas estructuras por imágenes. A su vez el desarrollo de sistemas de registro de la actividad eléctrica espontánea o inducida de esas estructuras permite definir con mayor exactitud su posición, límites, relaciones anatómicas con estructuras vecinas y comportamiento en relación al movimiento o a los síntomas componentes del parkinsonismo, lo que le confiere un carácter idóneo para establecer y caracterizar con seguridad el área afectada.
Todo este soporte tecnológico permite replantear la cirugía estereotáctica funcional como una opción terapeútica eficaz y segura. En ese contexto alrededor de 1985, se reinician las experiencias con dos resultados espectaculares; el control del temblor y la rigidez por el abordaje superselectivo del núcleo VIM del tálamo y la mejoría drástica de la hipocinesia con abolición de la toxicidad inducida por L-dopa por el abordaje de la región posteroventral del globus palidus medial.
En consecuencia existen suficientes evidencias para suponer que en los próximos años se impondrá el tratamiento quirúrgico como la primera opción terapeútica alternativa al uso crónico de la Levodopa.
Con una prevalencia estimada en alrededor de 1 a 2 por 1000 habitantes, aumentando esta cifra hasta un 4 por mil en la población mayor de 50 años y con una proporción superior al 50% de casos moderados-severos con complicaciones evolutivas o de casos caracterizados por pérdida evolutiva de la respuesta a la L-dopa y acentuada asimetría, podría estimarse la existencia de mas de 7.800 pacientes parkinsonianos susceptibles de tratarse por estos métodos en nuestro país, para quienes por otra parte pudiera no existir otra alternativa.
3.1.5- PROGRAMA DE EJERCICIOS
-
EJERCICIOS PARA MANTENERSE FÍSICAMENTE EN FORMA
Un adecuado programa de ejercicios es esencial para que el paciente pueda aumentar la movilidad, mejorar el equilibrio y la coordinación con el fin de mantener su independencia.
Los ejercicios que se describen a continuación, pueden ser útiles a la mayoría de enfermos de Parkinson. Sin embargo, es muy aconsejable diseñar un programa acorde con las necesidades de cada paciente. Por ello, su médico es quien mejor puede aconsejarle acerca de los ejercicios que más se adapten a su estado.
El programa de ejercicio puede ponerse en práctica de muy diversas maneras, dependiendo del paciente.
Cuando el paciente se fatiga con facilidad, se planificarán varias sesiones de corta duración a lo largo del día. Además, cada paciente debe familiarizarse con sus propias limitaciones para establecer su propio nivel de tolerancia al ejercicio.
Los pacientes que han estado inactivos durante largo tiempo, deberían empezar haciendo los ejercicios de una a cinco veces al día, para llegar progresivamente hasta diez o veinte veces diarias.
El primer grupo de ejercicios, es un programa inicial que incluye movilización y ejercicios generales para las principales partes del cuerpo. El segundo bloque, incorpora ejercicios para mejorar la coordinación y el equilibrio.
EJERCICIOS GENERALES
Ejercicios para la cabeza y el cuello.
Estos ejercicios están diseñados para proporcionar a los músculos y articulaciones del cuello un elevado grado de movilidad. Para realizarlos, siéntese en una silla y mantenga la espalda apoyada en el respaldo.
- Doblar la cabeza hacia adelante hasta tocar el pecho con la barbilla. Seguidamente, doble la cabeza hacia atrás hasta ver el techo. Girar la cabeza hasta ver el hombro derecho y luego hasta ver el hombro izquierdo.
- Ladear la cabeza hasta que la oreja derecha toque el hombro derecho (no levante el hombro).Colocar de nuevo la cabeza en posición vertical. Repetir el ejercicio hacia el lado izquierdo.
Ejercicios para los hombros.
- Encoger los hombros. Descanse. Empujar los hombros hacia atrás. Descansar. Repetir todo el ejercicio.
- Colocar las manos en la nuca y llevar los codos hacia atrás. Descansar.
Colocar las manos en las espalda tan arriba como pueda e intentar tocar sus omoplatos
Ejercicios faciales
Þ Elevar las cejas y arrugue la frente.
Þ Abrir la boca tanto como pueda.
Þ Hinchar los carrillos.
Þ Silbar.
Þ Mover la nariz.
Ejercicios respiratorios.
El objetivo de estos ejercicios, es mejorar la respiración.
- Colocar las manos sobre las costillas, coja aire y notar como se expansionan las costillas. Sacar el aire.
- Colocar las manos en el abdomen, coger aire y notar como se hincha el abdomen. Sacar el aire.
- Colocar las manos en el estómago, apretándolo, y expulsar el aire tan rápido como se pueda.
Ejercicios para los pies.
- Con los pies planos en el suelo, levantar alternativamente las puntas tan rápido como se pueda. Repetir el ejercicio pero elevar primero los talones y luego las puntas y así sucesivamente
-Sentado con la pierna levantada, mover el pie derecho describiendo círculos. Repetirlo con el pie izquierdo.
-Apoyado en algo seguro (por ej. un mueble), levantar los talones apoyándose en las puntas. Colocar de nuevo las plantas de los pies en el suelo y levantar luego las puntas apoyándose en los talones.
Ejercicios para las manos.
- Flexionar los brazos, mantener los codos al lado del cuerpo y las manos enfrente. Mover las muñecas describiendo círculos.
- Con los brazos en la misma posición que en el ejercicio anterior, abrir y cerrar las manos
Ejercicios para las piernas.
- Sentado en una silla, levantar la pierna hasta colocar el pie encima de un taburete situado enfrente. Inclinarse hacia delante y colocar las dos manos sobre la rodilla. Apretar hacia abajo para enderezar la rodilla. Mantenerse así y contar hasta 20. Descansar
- Sentado en una silla con los pies planos en el suelo, levantar la pierna izquierda y luego bajarla. Repetir el ejercicio con la pierna derecha.
- Apoyado en algo seguro (por ej. un mueble), levantar las rodillas como si caminara sin desplazarse.
- Apoyado sobre algo seguro (por ej. un mueble) separar una pierna de la otra.
- Levantar las rodillas y balancear los brazos como si caminara, pero sin desplazarse. La mano izquierda debe estar hacia adelante, cuando la pierna derecha esté elevada y viceversa.
EJERCICIOS PARA MEJORAR LA COORDINACIÓN DE MOVIMIENTOS Y EL EQUILIBRIO
Estos ejercicios le ayudarán en algunas de las dificultades más corrientes con que puede encontrarse.
Levantarse de una silla y sentarse.
- Situarse en el borde de la silla y apoyar bien los pies en el suelo. Separar los pies unos 20 ó 25 centímetros. Apoyar las manos en los brazos o borde de la silla. Inclinarse al máximo hacia adelante. Apretar hacia el suelo con los pies, empujar hacia adelante con los brazos y ponerse en pie.
- Si no lo consigue al primer intento, balancearse hacia adelante intentandolo de nuevo. Para sentarse, situarse lo más cerca posible de la silla, poner las manos en los brazos o bordes de la silla, inclinarse hacia adelante y luego sentarse.
- Cambiar la posición en la cama.
- Doblar las rodillas y apoyar los pies en la cama. Ladear las rodillas hacia la derecha.
- Entrelazar las manos y levantarlas estirando los codos.
- Girar la cabeza y los brazos hacia la derecha. Agarrar las manos al colchón para ajustar la posición hasta estar cómodo.
Marcha
- Caminar con los pies separados unos 15-20 centímetros.
- Levantar los pies como si marchara. Exagerar el balanceo de los brazos. Puede ayudarse llevando en cada mano una revista o un periódico enrollados. Procurar que los pasos sean lo más largos posibles. Es de ayuda caminar al ritmo de la música. Cuando gire, siga los consejos ya mencionados en el apartado media vuelta. Comprobar su postura en el espejo, para intentar corregirla.
Si nota que va a acontecer la festinación (propulsión o retropulsión) seguir los consejos mencionados en el apartado andar.
- No hablar mientras se marcha. Si se necesita decir alguna cosa, es mejor detenerse.
Corregir la postura.
- Ponerse de pie contra la pared tocándola con los talones.
- Tratar de mantenerse lo más recto y estirado que pueda, de modo que las paletillas y la parte posterior de la cabeza toquen la pared.
- Ponerse de pie de cara a la pared algo apartado de ella
- Estirarse hacia arriba todo lo que pueda con las palmas de las manos contra la pared. Mirarse las manos mientras se estira. Una vez estirado, mantener esta posición mientras se cuenta hasta 5.
3.2- SINDROME DE TOURETTE
3.2.1.- CONCEPTO E HISTORIA
En 1825 el primer caso de TS fue inscrito en la literatura médica con la descripción de la Marquesa de Dampierre, una mujer de la nobleza, cuyos síntomas incluían los tics involuntarios en muchas partes de su cuerpo y varias vocalizaciones incluyendo coprolalia y ecolalia. Ella vivió hasta los 86 años y su caso fue descrito por el Dr. George M. Gilles de la Tourette, el neurólogo francés que ha dado nombre a la enfermedad. Samuel Johnson y André Malraux fueron entre las personas famosas que se creé han sufrido el TS.
La Marquesa de Dampierre:

" ...a la edad de 7 años estuvo afligida por movimientos convulsivos de las manos y los brazos... Sintió que estaba sufriendo de sobreexcitación y travesuras, y... era objeto de reprimendas y castigos. Pronto se aclaró que éstos movimientos eran verdaderamente involuntarios... involucraba los hombros, el cuello, y la cara, y resultaba en contorsiones y muecas extraordinarias”.
Dr. George M. Gilles de la Tourette
El Síndrome de Tourette (TS) es un trastorno neurológico que se caracteriza por tics y demás; movimientos involuntarios, rápidos y repetidos que ocurren repetidamente de la misma manera. La causa no ha sido definitivamente establecida, el TS es originado por un tipo especial de hiperactividad en el cerebro. Teorías presentes apuntan a una sobreabundancia o sobreactividad de dopamina, es decir un metabolismo anormal de un componente químico que transmite mensajes neurológicos en el cerebro, también están involucrados otros neurotransmisores, actualmente se conoce la serotonina. Los tics pueden ser vistos como una manera normal del cerebro de " liberar vapor", bajo estas circunstancias.
En el ADD, parece haber una escasez de dopamina en otras partes "altas" del cerebro. Estas partes del cerebro parecen estar involucradas en suprimir estímulos insignificantes, en favor de los que realmente lo necesitan.
Los síntomas incluyen:
1. La presentación al mismo tiempo de tics motor múltiples y uno o más tics vocales en algún momento durante la enfermedad, aunque no necesariamente de manera simultánea.
2. La aparición de tics muchas veces al día (normalmente a brotes), casi a diario o de manera intermitente a lo largo de más de un año.
3. Los cambios periódicos en el número, frecuencia, tipo y localización de los tics, y también en su intensidad, los síntomas pueden hasta desaparecer durante varios meses seguidos.
4. El comienzo de la enfermedad ocurre antes de los 21 años.
Además:
-
Usualmente tiene una temprana aparición en la niñez
-
Es hereditaria
-
Es neurológica, y no psicológica
-
Afecta más a varones que a hembras
-
No empeora de manera progresiva
Por lo usual, los primeros síntomas de ST son tics de la cara - más comunmente parpadeo. Además, los tics de la cara pueden incluir contracción de la nariz o muecas. Con el tiempo, otros tics motores aparecen, tales como sacudir la cabeza, extender el cuello, patear o retorcer y doblar el cuerpo.
A menudo, los pacientes de ST emiten sonidos, palabras, o frases raros e inaceptables. Es común que la persona con ST continuamente aclare la garganta, tose, gruñe, olfatee, ladre, grazne o grite.
A veces, las personas con ST gritan obscenidades o groserías involuntariamente (coprolalia) o repiten las palabras de los demás constantemente (ecolalia). A veces, tocan a otras personas excesivamente o repiten acciones obsesiva e innecesariamente. Algunos pacientes con ST severo muestran conducta automutilante como morderse los labios o la mejilla y golpearse la cabeza contra objetos duros. Sin embargo, estas conductas son extremadamente raras.
Los tics aumentan y disminuyen de severidad alternadamente y de vez en cuando cambian en número, frecuencia, tipo, y localización. Los síntomas pueden apaciguarse por semanas o meses para luego reaparecer más tarde.
En algunas ocasiones el TS provoca burlas y rechazo par la sociedad, vecinos, profesores, y hasta uno que otro observador. Los padres se pueden sentirse agobiados por las rarezas del comportamiento de sus hijos. El niño puede ser amenazado, excluido de las actividades familiares, e impedirle el disfrute de una relación personal normal. Estas dificultades pueden llegar a ser más graves durante la adolescencia, una especial de prueba para las personas jóvenes y aún más para una persona que esté padeciendo un trastorno neurológico. Para evitar daños psicológicos, el tratamiento y diagnóstico temprano son cruciales. Más aún, en casos graves es posible controlar los síntomas con medicación.
Muchas personas experimentan una completa remisión o una marcada mejoría a finales de la adolescencia o cuando cumplen los veinte y pocos años. La mayoría de personas con TS mejoran, no empeoran, según van madurando, y pueden anticipar que llevarán una vida normal. Aproximadamente un tercio de los pacientes experimentan una disminución marcada de los tics en la edad adulta.
3.2.2- LOS TICS
Clasificación de los tics
Existen dos categorías de tics: simples y complejos. Los tics simples son breves movimientos repentinos que involucran un número limitado de grupos de músculos. Estos ocurren de una manera singular o aislada y a menudo se repiten. Algunos de los ejemplos más comunes de tics simples incluyen parpadear, encoger los hombros, ceñir el entrecejo, sacudir la cabeza, graznar, y olfatear. Los tics complejos son distintos modos coordinados de movimiento sucesivos involucrando varios grupos musculares. Los tics complejos son:
-
Motores; saltos, tocar a las demás personas o cosas, olfatear, dar giros, y rara vez actos de autolastimarse, incluido el golpearse o morderse.
-
Vocales; la expresión de vocabulario o frases fuera de contexto, y coprolalia (el empleo de palabras obscenas en público).
Algunos otros son: brincos en el ojo; comer uñas; toser; silbar; zumbar; tartamudear; súbitos cambio del tono de la voz, velocidad o volumen.
La variedad de tics o síntomas parecidos a los tics que se pueden encontrar en los TS es enorme. La complejidad de algunos síntomas a menudo confunden a los miembros de la familia, amigos, maestros, y empresarios, quienes pueden encontrar difícil de creer que las acciones o las palabras emitidas sean involuntarias.
Se percibe que el decir malas palabras, a menudo suele ser el aspecto más penoso y dramático del TS y es por eso que ha recibido el término médico de coprolalia.(latin: labios de heces).
Los tics son aparentemente al azar
Los hábitos son maneras permanentes de ejecutar acciones que, solamente se utilizan cuando son necesarios. Usted puede tener un manera especial de revolver el café pero lo hace, después de todo, sólo mientras revuelve su café. A menos que haga un esfuerzo para olvidarlos, los hábitos serán parte de usted de por vida.
En contraste, el impulso del tic puede aflorar en cualquier hora de día, aparentemente, sin relación a cualquier otra cosa que usted este haciendo en ese mismo período. También los tics inesperadamente después de meses o años de frustrado intento por "parar de hacerlos", sistemáticamente desaparecen y son reemplazados por otros tics.
La magnitud a la que se experimentan los tics es al azar, varia de individuo a individuo. Muchos investigadores creen que son totalmente casualidad y sin sentido "el nervio tira bruscamente", mientras muchas personas quienes realmente padecen del TS, informan que parece haber algun patrón o significado escondido, que no pueden entender de ellos.
Los tics son impulsos fuertes que requieren expresarse
Los tics son como una sensación irresistible, como la necesidad de estornudar o rascarse la picadura de un mosquito, y que finalmente el paciente tiene que realizar. Las personas con el TS a menudo buscan un lugar retirado donde dar rienda suelta a sus síntomas después de haberlos estado aguantando durante las horas escolares o de trabajo. Típicamente, los tics aumentan como resultado de una tensión o presión y mejoran con la relajación o la concentración sobre un trabajo absorbente.
Algunos touréticos pueden, consciente o inconscientemente suprimir sus tics desde minutos, segundos y hasta horas. Sin embargo, entre más tiempo sean controlados, mayor es la tensión interna y agitación que tarde o temprano provoquen de nuevo su aparición.
Una manera de verificar si una acción es un tic o no, es el de aplicar la prueba de "tratar de suprimirlo" . Si lo suprime y lo lleva a una agitación, eventualmente hace el acto u otra acción similar, o un sentimiento de "porqué debo suprimirlo yá - quiero hacerlo de todos modos," lo más probable es que sea un tic.
Premoniciones y conocimiento de tics repentinos
Algunos touréticos informan que están conscientes del impulso del tic antes de realizarlo. En ese caso, el impulso puede expresarse como un recuerdo fugaz que usted tiene este tic y quiere hacerlo, una idea súbita o imagen de frustración que parece querer expresar en el tic, o como la idea de que los tics aumentan en usted como lo hace un estornudo.
Algunas personas con TS están conscientes que el tic es un conjunto de sensaciones físicas y emocionales desagradables, que buscan salir "al mundo".
Los tics parecen no tener ningún propósito
Considerando que los impulsos normales tales como "rascarse una comezón" y toser, tienen un propósito, los tics exteriormente no parecen tener ninguno. Por ejemplo la diferencia entre el decir malas palabras de manera natural y la coprolalia, es que un tourético a menudo ni siquiera se enoja o agita en lo absoluto cuando el insulta y a menudo esta acción emerge totalmente fuera de contexto.
Para resumir: en acciones normales, usted decide hacer algo, como levantarse y salir. Después de la decisión y sólo si desea hacerla, usted realmente la ejecuta.
Con los tics es casi lo contrario: no hay premeditación (pensar) en hacerlos y sólo si usted decide suprimir el tic (y es capaz de hacerlo), usted acaba por no experimentar la acción - durante algún tiempo.
Si desea imaginarse como actúan los tics: imagiínese que está consciente del estímulo de estornudar sin ninguna sensación física en su nariz antes de hacerlo. Ahora reemplace "estornudar" con cualquier otra acción repentina que pueda pensar.
O imagine la acción de cerrar sus ojos porque una mosca vuela directamente hacia ellos, pero sin que haya realmente ninguna mosca. Ahora imagínese que cuando se resiste al impulso de cerrar sus ojos, la mosca "imaginaria" se congela delante del globo de sus ojos para siempre, desesperantemente buscando entre ellos hasta que eventualmente usted tiene que cerrar sus ojos o dar una sacudida para alejarla de su cara.
3.2.3- Ecolalia, Ecopraxia, Palilalia, y otras repeticiones
En relación a los tics, hay otros estímulos:
-
Ecolalia, el impulso de repetir palabras de otra persona.
A veces en conversaciones, palabras insignificantes o fin de frases, que de algún modo captan su atención y usted se siente obligado a repetirlos...
-
Ecopraxia, el impulso a imitar acciones de otra persona
Usted se encuentra repitiendo cada arrastre del pie, o bien, camina detrás de alguien y se halla imitando su andado.
-
Palilalia, el impulso de repetir sus propias palabras o pensamientos
Usted encuentra que esas personas alrededor suyo, a menudo piensan que les está hablando a ellas; pero es solo que usted mantiene y externa sus pensamientos en voz alta...
-
Otras repeticiones Muchos touréticos informan que a veces se hallan ellos mismos en un círculo repetitivo de acciones o pensamientos. A menudo ellos sienten que la única manera de acabar con esas repeticiones es con un tic.
Cuando mis amigos y yo vamos a algún lado, a menudo me encuentro repitiendo palabras de leí fuera, en voz alta. La conversación es algo como:
"Lavandería automática."
"Qué?"
"Oh, nada."
c) Coprolalia, o decir malas palabras
Éste es generalmente el más conocido y dramático aspecto del TS. Cuando el TS captó la atención de los medios de comunicación en el pasado, era principalmente debido a este aspecto tan sensacional del síndrome. Títulos llamativos como "Maldita Enfermedad" o "El Síndrome Malhablado" fueron usados. Con tristeza, muchas veces es el único aspecto del TS que normalmente se conoce (y que es objeto de burla.)
La verdad es que el decir malas palabras no es un síntoma universal del TS. La coprolalia ocurre en aproximadamente de 8 a 30 por ciento de los casos, y con frecuencia permanece sólo en una fase de la vida de la persona.
La coprolalia puede ser uno de los más angustiosos y aun así fascinantes aspectos del TS. Muchos de los que muestran coprolalia viven en ambientes donde el decir palabras obsenas no era permitido o perdonado. Muchos no se perdonan a si mismos por el hecho de estar insultando. La coprolalia puede también causar problemas sociales, produciendo la falsa impresión que esa persona ofende a otras.
Asimismo, es por esta razón que los touréticos muchas veces y de manera errónea, han estado pensando que padecen de una deficiencia moral, y que el TS es un transtorno psicológico.
La pregunta que queda es: ¿cómo puede una condición biológica, a menudo salir a la superficie como una predisposición a pronunciar palabras ciertamente obscenas? Puedo proponerles dos explicaciones:
Si considera que decir palabras obscenas es reprobable, o si usted mismo se insulta, las malas palabras a menudo llevan un valor emocionalmente fuerte - ese es su propósito, después de todo. Aunque se especifica que la forma en que se determinan las malas palabras es mediante un análisis cultural y no biológico, es posible que, cuando el individuo crezca y aprenda su significado, estas se encuentren "guardadas" en relación con ciertos sistemas emocionales específicos del cerebro.
Hay malas palabras que son más preferidas que otras. Parece como si las palabras ofensivas más comunes, tuvieran cierta calidad sonora que les diera cierta agudeza; tal como consonantes explosivas o repetición de fonemas (ejemplos de fononemas: mata y bata, sal y sol, etc). Estos mismo aspectos las hacen ideales para el tic. De hecho, otros tics vocales frecuentemente parecen llevar una cierta calidad rítmica.
3.2.4- Problemas Emocionales, de Comportamiento y otros Relacionados
Las personas con el TS presentan además de los tics otros comportamientos asociados, como son:
Obsesiones que consisten en repeticiones indeseadas de pensamientos molestos.
Compulsiones y Conducta Ritualista, por lo que las personas sienten que deben hacer algo una y otra vez o hacerlo de una cierta forma. Ejemplos incluyen tocar un objeto con una mano después de haberlo tocado con la otra, sólo para "equilibrar las cosas" o revisar repetidamente si el fuego de la cocina está apagado. Los niños a veces les ruegan a sus padres que repitan una frase muchas veces hasta que "suene bien".
Trastorno de Déficit de la Atención, con o sin Hiperactividad (ADD o ADHD), que ocurre en muchas personas con TS. A menudo los niños muestran signos de hiperactividad antes de que aparezcan los síntomas del TS. Síntomas de hiperactividad y ADD pueden incluir: dificultad de concentración; no terminar lo que empezó; aparentar que no se escucha; ser fácilmente distraible; actuar a menudo sin pensar; cambiar constantemente de una actividad a otra, necesitar mucha atención, e intranquilidad en general. Los adultos pueden tener signos residuales de ADHD tales como un comportamiento impulsivo y dificultades en concentrarse, y la necesidad de moverse constantemente. ADD sin hiperactividad incluye todos lo síntomas mencionados arriba el alto nivel de actividad. A medida que los niños con ADHD crecen, La necesidad de moverse se expresa por intranquilidad y conducta inquieta. Las dificultades con la concentración y el deficiente control de los impulsos persiste.
Incapacidad de aprendizaje, tales como dislexia, trastornos aritméticos, y dificultades perceptivas.
Dificultades con el control de impulsos, que pueden resultar en casos raros, en comportamientos de agresividad excesiva o actos inapropiados socialmente.
Trastornos del sueño, los cuales son bastante comunes en personas con TS. Entre éstos se incluyen despertares frecuentes y el caminar o hablar en sueños.
a) Transtorno Obsesivo-Compulsivo (OCD)
Entre algunos síntomas comúnes del TS se encuentran como se mencionó anteriormente, las obsesiones y obligaciones (compulsiones), las cuales van en la clasificación de Transtorno Obsessivo-Compulsivo (OCD). Uno podría ver el segundo como tics sumamente complejos.
Las obsesiones son ideas recurrentes (que se repiten) o pensamientos que parecen invadir y toman mando de su mente, sin su consentimiento. El volumen de las obsesiones puede variar. Ejemplos de estas son:
-
obsesiones sobre la violencia y reiteradamente imagina escenas violentas
-
obsesiones con respecto a los números y cuantifica todo (contar)
-
obsesiones sobre las palabras y ortografía
-
obsesivamente tiene que preguntar todo
Las obligaciones (compulsiones) son acciones repetitivas, disposiciones que uno se siente obligado a hacer, con frecuencia de una manera ritualista. Frecuentemente estos actos se van hasta el término a pesar del hecho de que no quiere verdaderamente hacerlos, y a pesar de querer resistirlos. Ejemplos de estos son:
-
tener que hacer cosas pequeñas ahora mismo
-
siempre alinea todos los libros perpendicularmente con la mesa
-
tener que salir de la misma manera en que entró
-
revisar y volver a verificar "chequear" algo
Uno podría decir que las obligaciones son lo mismo que los tics: un impulso similar está presente, pero, no se llevan a cabo automáticamente porque son demasiado largos y complejos; estos son llevados hasta su término de una forma deliberada, como uno finalmente lo tiene que poner en práctica.
b) Transtorno de Déficit de la Atención (ADD)
El Transtorno de Déficit de la Atención (ADD) es uno de los compañeros más comúnes del TS - tanto como un 50% de los touréticos tienen algo de él.
En la mayoría de casos, el ADD se acompaña por hiperactividad, que lleva al nombre de Trastorno de Déficit de la Atención con Hiperactividad, (ADHD). En años anteriores las personas hablaron de "niños hiperactivos." Lo que menos se sabe es que estos niños pueden crecer y ser ADD adultos.
Los principales síntomas del ADD son la falta de atención y de impulsividad - dificultad para mantener enfocada su atención en una cosa, y ser susceptible a un amplio rango de distracciones.
Tempranamente en vida de muchas personas que padecen ADD, experimentan problemas en la escuela y es por esta razón que sus padres a menudo buscan ayuda. La norma parece ser que, la mayoría de touréticos que son diagnosticados en la niñez o la adolescencia sufren de "Síndrome de Tourette con ADD". Si la formación médica de su doctor desconoce el diagnóstico del TS o si el está poco familiarizado con sus formas más apacibles (menos severas), puede que nunca se llegue al diagnóstico del TS.
c) Caprichos, Episodios y Sentimientos Singulares
Muchos touréticos informan que están inclinados a caprichos tal como el desánimo, y sentimientos singulares tal como que el mundo es sólo una película o que allí hay algo terriblemente importante en el contorno específico del sacapuntas de su escritorio.
La principal característica de todo ésto parece estar que de repente aparecen y desaparecen "inesperadamente " de la misma manera. También, de una forma racionalmente astuta, estos engaños no siempre ayudan - una parte más honda de la mente parece haber encontrado su sustento en ellos. Sólo aparecen nuevamente sin previo aviso.
3.2.5- TRATAMIENTO
Por el hecho de que los síntomas no limitan a la mayoría de los pacientes y su desarrollo procede normalmente, la mayoría de las personas con ST no requieren medicamentos. No obstante, hay medicamentos disponibles para ayudar a los pacientes cuando los síntomas interfieren con las tareas cotidianas.
Lamentablemente, no existe un sólo medicamento útil para toda persona con ST. Asimismo, no hay un medicamento que elimine todos los síntomas y todos los medicamento tienen efectos secundarios. Además, los medicamentos disponibles para el ST solamente puede reducir síntomas específicos.
Algunos pacientes que necesitan medicamentos para reducir la frecuencia e intensidad de los tics pueden ser tratados con fármacos neurolépticos como haloperidol y pimocida. Se administran estos fármacos usualmente en dósis muy pequeñas las cuales se aumentan lentamente hasta que se logra el mejor balance posible entre los síntomas y los efectos secundarios.
Recientemente, los científicos han descubierto que el uso de fármacos neurolépticos a largo plazo pueden causar un trastorno de movimiento involuntario que se llama discinesia tardía. Sin embargo, esta condición usualmente desaparece al descontinuar el medicamento. Los efectos secundarios a corto plazo de haloperidol y pimocida incluyen rigidez muscular, babeo, temblores, falta de expresión facial, movimiento lento y desasosiego. Estos efectos secundarios pueden reducirse mediante fármacos usados comúnmente para tratar la enfermedad de Parkinson. Otros efectos secundarios como fatiga, depresión, ansiedad, aumento de peso y dificultad en pensar claramente pueden ser más molestos.
La clonidina, un fármacoantihipertensivo, también se usa para tratar los tics. Las investigaciones muestran que este fármaco es más eficaz para reducir los tics motores que los tics fónicos. Los efectos secundarios comunes asociados con el uso de clonidina son fatiga, sequedad bucal, irritabilidad, mareos, dolores de cabeza e insomnio. Flufenacina y clonacepam pueden recetarse para ayudar a controlar los síntomas de los tics.
También hay medicamentos disponibles para tratar algunos de los trastornos de conducta asociados con el ST. Estimulantes tales como metilfenilato, pemolina y dextroamfetamina, usualmente recetados para el trastorno de déficit de la atención, son algo efectivos pero su uso es controversial porque se ha reportado que éstos aumentan los tics. Para las conductas obsesivo-compulsivas que significativamente interfieren con el funcionamiento cotidiano se puede recetar fluoxetina, clomipramina, sertralina y paroxetina.
Otros tipos de terapia pueden ser útiles. A pesar de que los problemas psicológicos no causan el ST, la psicoterapia puede ayudar a la persona a manejar no sólo el trastorno sino también los problemas sociales y emocionales que ocurren a veces.
Las técnicas de relajamiento y bioretroalimentación pueden ser útiles para aliviar el estrés que puede provocar un aumento de los síntomas de los tics.
3.2.6- HERENCIA Y TS
Los estudios genéticos indican que el TS es hereditario a través de un gen dominante que puede producir síntomas distintos en diferentes miembros de una familia con TS, y que existe un cincuenta por ciento de probabilidades de transmitir el gen a alguno de sus hijos. No obstante, ese gen que se hereda puede manifestarse como el TS o también en forma de tics leves o con rasgos de obsesión compulsiva sin ningún tic. Actualmente se conoce que existe una incidencia más alta de lo normal de trastornos de tics leves y comportamientos obsesivo-compulsivos en las familias de pacientes con TS.
El sexo del niño también determina la expresión del gen. Las probabilidades de que un niño padezca un trastorno característico de las personas con TS, es de, por lo menos, tres veces más alto en el varón que en la hembra. Aún así, sólo alrededor del diez por ciento de los niños que heredan el gen tendrían síntomas lo suficientemente graves como para recibir tratamiento médico. En ciertos casos, el TS podría no ser hereditario y se identifica como TS esporádico. La causa es desconocida en estos casos.
3.2.7- investigaciones recientes
Dentro del gobierno federal de USA, el apoyo más importante para las investigaciones sobre el ST y otros trastornos neurológicos proviene del Instituto Nacional de Trastornos Neurológicos y Apoplejía—National Institute of Neurological Disorders and Stroke (NINDS). El NINDS es parte de los Institutos Nacionales de Salud—National Institutes of Health (NIH)—y es responsable de apoyar y llevar a cabo investigaciones científicas del cerebro y el sistema nervioso central.
El NINDS patrocina las investigaciones del ST no sólo en los laboratorios del NIH sino también proveyendo fondos de investigación a las instituciones médicas más importantes del país. El Instituto Nacional de Salud Mental, el Centro Nacional de Recursos para la Investigación, el Instituto Nacional de Salud Infantil y Desarrollo Humano, el Instituto Nacional sobre Abuso de Drogas y el Instituto Nacional de Sordera y otros Trastornos de Comunicación apoyan también investigaciones relacionadas al ST.
Por otra parte, desde 1984, La TSA (Asociación del Síndrome de Tourette) ha subvencionado directamente investigaciones importantes en un gran número de áreas científicas relevantes para el TS. Recientemente, los estudios se han intensificado para poder comprender cómo el trastorno se transmite de una generación a la siguiente, y varios investigadores están trabajando en el camino de conocer la localización del marcador genético del TS. Ese enfoque ha sido reforzado por los esfuerzos de un grupo de apoyo de científicos internacionales del TS, quienes han formado una única red para compartir lo que se conoce sobre la genética del TS y su colaboración sistemática para descubrir lo desconocido. Se está obteniendo información adicional con los estudios de grandes familias con numerosos miembros que padecen el TS. A su vez, los investigadores continúan con su estudio específico sobre grupos de componentes químicos del cerebro para mejorar la comprensión del síndrome y para identificar nuevos y mejores medicamentos.
Investigaciones recientes han conducido a varios avances notables en el entendimiento del ST. Los científicos han aprendido que el ST es heredado de un gen o genes dominantes que causan síntomas distintos de paciente a paciente, y que el trastorno es más común de lo que se pensó previamente.
Estudios genéticos. Actualmente, los investigadores están llevando a cabo estudios de eslabonamiento genético en familias multigeneracionales grandes afectadas con ST en un esfuerzo por encontrar la localización cromosomal del gen o genes del ST. El encontrar un marcador genético (una anormalidad bioquímica que todos los pacientes con ST compartan) sería un paso de importancia para entender los factores de riesgo genéticos del ST. Una vez se encuentre el marcador, los esfuerzos de investigación podrían enfocarse en localizar el gen o genes del ST.
El entender la genética del ST beneficiaría a los pacientes que se preocupan de una recurrencia en sus familias y ayudaría a aclarar el desarrollo del trastorno. El localizar el gen o los genes del ST fortalecerá la diagnosis clínica, mejorará la consejería genética, aclarará la patofisiología y proveerá pistas para terapias más eficaces.
Estudios de neurotransmisores. Los investigadores siguen investigando algunos neurotransmisores para aumentar nuestro entendimiento del síndrome, explorar el papel que juegan en el proceso de la enfermedad y proveer terapias más eficaces.
Estudios ambientales. Otros proyectos de investigación actualmente en progreso incluyen el análisis de niños de alto riesgo no afectados con el propósito de indentificar factores ambientales, tales como el estrés de la vida o el uso de medicamentos, que pueden influenciar la expresión del trastorno.
Los científicos están llevando a cabo pruebas neuropsicológicas y estudios de neuroimágenes de la actividad y estructura cerebral para determinar a que grado exposiciones ambientales específicas pueden afectar el surgimiento de los tics y/o síntomas obsesivo-compulsivos.
3.3- ESQUIZOFRENIA
3.3.1- INTRODUCCIÓN
Las psicosis son trastornos funcionales u orgánicos del sistema nervioso que constituyen desviaciones graves de la normalidad mental. Se caracterizan por cambios profundos en la personalidad. Tanto el modo de pensar como el de razonar, juzgar y querer, sufren radicales transformaciones, es decir, existe una desorganización transitoria o definitiva de la personalidad.
La esquizofrenia es la psicosis por excelencia. Pese a los indiscutibles adelantos en el conocimiento de la enfermedad y en su tratamiento, continúa siendo la patología que provoca el mayor número de ingresos a instituciones para enfermos mentales de todo el mundo, y aún es vista como un verdadero “cáncer” de la mente.
Para explicar los diversos cuadros clínicos de la patología esquizofrénica se han utilizado fundamentalmente dos modelos bioquímicos:
- El anfetamínico, con su aplicación a la disrregulación de la neurotransmisión dopaminérgica, y que estaría relacionado con la sintomatología paranoide;
- El de la fenciclidina (PCP) que estaría relacionado con la sintomatología de tipo catatónico.
Otros modelos recientes se refieren a la disfunción de los sistemas inhibidores moduladores de la neurotransmisión del ácido Gamma Amino Butírico (GABA) y de la glicina.
La actividad dopaminérgica en el Sistema Nervioso Central tiene roles funcionales claramente diferentes, según la estructura neuronal en donde se asientan. Así, pues, recordemos las cuatro vías dopaminérgicas implicadas en la fisiopatología de la enfermedad:
- La vía mesocortical: su disfunción es posiblemente la causa principal de los síntomas negativos y del deterioro crónico de los pacientes;
- La vía mesolímbica: es la responsable de los síntomas positivos delirantes alucinatorios;
- La vía nigroestriada: se relaciona con los efectos extrapiramidales;
- La vía tuberoinfundibular: relacionada con los síntomas endócrinos.
Los múltiples estudios neuroanatómicos y neuroquímicos referentes a la patología esquizofrénica, así como los estudios de localización funcional en el cerebro de pacientes con esta patología, nos impone la necesidad de establecer un modelo conceptual no unitario de lo que hoy llamamos esquizofrenia.
La correlación entre la clínica y las condiciones biológicas deberá ubicarse dentro del contexto de un amplio espectro de diferentes manifestaciones psicopatológicas, que en forma didáctica más que real y de acuerdo con las tendencias actuales, tendrían expresión entre las tendencias tipo I (agudas) y las esquizofrenias tipo II (crónicas).
La experiencia clínica nos indica que en un porcentaje importante las manifestaciones psicopatológicas se pueden categorizar como correspondientes al tipo I o II de esquizofrenias, pero igualmente existe una categoría intermedia que de acuerdo con la teoría de los campos borrosos, no es lo uno ni lo otro y es ambas a la vez
3.3.2- Clasificación de la esquizofrenia
Es bien conocida la propuesta de Crow (1980) de clasificar la esquizofrenia en subtipos I y II.
- subtipo I, de cuadro clínico agudo, con síntomas “positivos”, como alucinaciones, delirios, trastornos del pensamiento, sin evidenciar trastorno intelectual, con buena respuesta a los antipsicóticos convencionales y a los atípicos, y cuya alteración neuroquímica sería una hiperactividad D2.
- subtipo II, de curso crónico, con síntomas “negativos”, como aplanamiento afectivo, pobreza del discurso, abulia, amimia, en ocasiones con trastorno intelectual, con una pobre respuesta a los antipsicóticos convencionales, pero con una mejor repuesta a los antipsicóticos atípicos, cuya alteración neuroquímica está dada por una disrregulación DA-5HT2, y donde existe destrucción celular y alteraciones estructurales del cerebro. La presencia de signos neurológicos es de particular interés. Incluyen las discinesias tardías con la implicación de que son efectos tardíos de la administración neuroléptica pero que también se han visto antes de la introducción de tal medicación y están presentes en pacientes que nunca han recibido tales drogas. Esos movimientos están más relacionados con manifestaciones tardías de la enfermedad que a su tratamiento, por lo que los incluye como una parte del síndrome tipo II. El estado defectual o síndrome tipo II es relativamente no- respondiente a la medicación neuroléptica y está muy relacionado con los deterioros crónicos de la esquizofrenia. El correlato clínico de cambio estructural en el cerebro ha sido variable pero cuando se presenta tiende a estar entre los rasgos del síndrome tipo II. Algunas evidencias de investigación postmortem sugieren que el tipo II está asociado con pérdida de CCK de las neuronas de las regiones límbicas y una reducción de MAO-B (que puede localizarse en las células gliares). La edad de comienzo es importante: los pacientes con un comienzo más temprano son más propicios a desarrollar síntomas negativos y deterioro intelectual que aquellos de comienzo más tardío. Se evidencia una relación entre cambio estructural y función cognitiva. Algunos hallazgos sugieren que la esquizofrenia podría considerarse como una anomalía del desarrollo, más que un proceso degenerativo atribuible a una noxa exógena
3.3.3- Consideraciones neuroanatómicas e inmunohistoquímicas
Los hallazgos neuroanatómicos son diversos, pero se encuentran sobre todo en pacientes de tipo II y en el hemisferio izquierdo o dominante. Los hallazgos de Benes y col., unidos a la correlación neuropatológica en esquizofrenia, aparentemente confirman la impresión de defectos estructurales en corteza, cerebelo, sistema límbico, etcétera, 1que podrían ser la consecuencia de los cambios biológicos en la esquizofrenia.
Existe, según algunos autores, una disminución de la capa III pre-frontal, una disminución de la capa IV de la corteza cingular, una disminución en la capa III de la corteza premotora. Estas áreas reciben abundantes aferencias del tracto dopaminérgico mesocortical del área tegmental ventral, y juegan un papel importante en funciones como la atención, el humor y la conducta, las mismas que están afectadas en los procesos esquizofrénicos.
Las investigaciones neuropsicológicas apuntan a una hiperfunción frontal asociada a una disfunción del lóbulo temporal antero-medial que altera las vías responsables de la memoria de trabajo, por alteración de las vías monoaminérgicas ascendentes.
Se postula una modulación cortical de la actividad dopaminérgica subcortical, aunque esto es aún incierto.
Las investigaciones con técnicas de neuroimagen son de gran relevancia en el estudio de las esquizofrenias, aunque todavía no resultan lo suficientemente concluyentes, tal vez por no existir una clara propuesta de diferenciar los casos de investigación de acuerdo con el predominio de las manifestaciones psicopatológicas personales o familiares del paciente. Por esto, los resultados son comparables a los que se obtienen en otros problemas no esquizofrénicos, y aun con los de muestras comparativas con poblaciones normales. Sin embargo, pensamos que en el futuro la investigación de tipo longitudinal nos permitirá respuestas más válidas y coherentes.
De entre las técnicas de imagen, destaca en su perspectiva futura la Resonancia Magnética-Espectroscópica, cuyos primeros reportes publicados por Jay E. Pettegrew, en el Arch. Gen. Psychiatry, 1991, demuestran alteraciones en la membrana fosfolipídica en la región dorsal del córtex pre-frontal de pacientes durante el primer episodio esquizofrénico. Este estudio concluye con la verificación de que los pacientes esquizofrénicos examinados presentan una significativa reducción de los niveles de fosfomonoesteres y ortofosfatos inorgánicos, y un significativo incremento de los niveles de fosfodiesteres y trifosfato de adenosina.
Esto indicaría que existe un disbalance entre los procesos degenerativos y restauradores de las membranas neuronales, proponiéndose que existiría un déficit en la enzima fosfodiesterasa, encargada de la conversión de fosfodiesteres en fosfomonoesteres en las membranas celulares. Estos datos relevantes permiten especular que existiría una tendencia precoz neuro-degenerativa que conduciría a la hipoactividad funcional de estas regiones cerebrales.
El trabajo de investigación se realiza con dos tipos de cuadros esquizofrénicos: el tipo paranoide y el tipo indiferenciado, esto es, el que nosotros hemos manifestado debe ubicárselo en la mitad del espectro clínico de las esquizofrenias.
Estudios longitudinales con técnicas de imagen, de personas con predisposición heredo-familiar, durante y después de la primera crisis, y en correlación con la evolución clínica particular de cada caso, podrán en un futuro entregarnos resultados más fiables que los estudios de tipo transversal.
Los estudios de inmunohistoquímica del doctor Schahram Akbarian, del Departamento de Anatomía y Neurobiología de la Universidad de California, en Irvien, proponen la hipótesis de una alteración migratoria neuronal primitiva. Se ha identificado un grupo de neuronas corticales que contienen la enzima nicotin-adenin-dinucleótido-diaforasa (NADPH-d), que cataliza la oxidación de NADPH a NADP, y que aparentemente condiciona una resistencia particular a los procesos de neurodegeneración o neuronecrosis.
Normalmente estas neuronas se ubican en las capas más externas de la corteza frontal y temporal, y en los esquizofrénicos se halla la mayor concentración unos milímetros más abajo, inmersas en la substancia blanca, lo que fisiológicamente llevaría a una alteración de las conexiones frontales y temporales, cuya expresión serían los síntomas esquizofrénicos. La migración anómala podría ser consecuencia de afecciones virales de la madre durante el embarazo.
La enzima NADHP-d tiene una acción protectora contra el óxido nítrico y radicales libres oxigenados, elementos que tienen que ver con procesos neurotóxicos y neurodegenerativos.
3.3.4- Consideraciones neuroquímicas
Se postula en la esquizofrenia la existencia de una alteración de la función dopamínica y serotonínica, así como de otros neurotransmisores y neuropéptidos como la noradrenalina, el glutamato, el GABA, la colecistokinina, la neurotensina y la meta-encefalina.
Se ha relacionado la dopamina y los receptores dopaminérgicos con los síntomas que caracterizan a las esquizofrenias tipo I y II. Una actividad alta de dopamina en las regiones cerebrales A 10, y niveles altos de ácido homo vanílico (HVA) en plasma, corresponde a los estados psicóticos agudos o de esquizofrenia tipo I. Una baja actividad de dopamina en la región prefrontal con disminución del flujo cerebral regional, corresponde a los estados deficitarios o de esquizofrenia tipo II.
La contraposición aparente de estados hipodopaminérgicos e hiperdopaminérgicos en la explicación de los síntomas esquizofrénicos es más aparente que real, puesto que experimentalmente se ha podido comprobar en ratas que lesiones de las neuronas pre-frontales de la corteza producen como respuesta un aumento en el estriado de dopamina, HVA y DOPAC (ácido dihidroxi-fenil-acético) y un aumento de receptores tipo D2 de dopamina.
En cambio si se inyecta en regiones pre-frontales el agonista dopaminérgico apomorfina, se produce una notable disminución en el estado de HVA y DOPAC.
La implicación que tiene la señal nor-adrenérgica en este fenómeno parece ser importante, ya que si se realizan lesiones que toman por igual las vías DA y HA, la activación superficial no se presenta.
En seres humanos, cuando ocurren lesiones por apoplejía en las regiones corticales, se produce una activación de la actividad dopaminérgica subcortical en el lado de la lesión.
Los receptores de dopamina que se conocen hasta hoy son:
- D1: acoplados a la actividad de la adeniciclasa y localizados en la corteza de seres humanos;
- D2: asociación negativa a la adenilciclasa. Se reconocen el D2a y el D2b, y se localizan preferencialmente en el sistema límbico y estriado;
- D3: de localización mesolímbica;
- D4: en asociación con el receptor del nucleótido Guanina, y de amplia distribución mesolímbica. Se han identificado las variaciones polimórficas D4-2, D4-4 y D4-7, y es muy probable la existencia de las variaciones D4-6 y D4-8. Este receptor D4 tiene gran afinidad con el antipsicótico clozapina.
- D5: de localización mesocortical y de gran afinidad dopaminérgica, con activación de la enzima adenilciclasa.
Desde hace algunos años se ha planteado la hipótesis del importante rol que tienen las endorfinas en la modulación de la actividad dopaminérgica, las mismas que ejercerían su acción a nivel pre-sináptico, acoplándose a receptores que activan una serie de sucesos bioquímicos que tienen que ver con la fosforilación de las proteínas.
Hay un modelo que explica la enfermedad de Parkinson, consistente en la acción de la neurotoxina MPTP (1 Metil-4 fenil 1-2-3-6 Tetrahidro-piridina) que injuria a neuronas de tipo dopaminérgico. Ésta es una molécula hidrofóbica que atraviesa fácilmente la barrera hematoencefálica y que por acción de la IMAO-B se convierte en un metabolito activo: el MPP (N-Metil-4 Fenil Piridium).
Este metabolito es transportado por la membrana plasmática por medio de proteínas transportadoras que utilizan sodio en la co-transportación fisiológica de monoaminas y otras moléculas pequeñas.
Se ha demostrado que la neuronas del tegmento ventral son más resistentes a la injuria del MPTP que las neuronas de la substancia negra. Esta diferencia de vulnerabilidad se aprecia entre poblaciones celulares dopaminérgicas y entre diferentes especies de animales.
El transportador de membrana en general tiene especificidad para las diferentes monoaminas. Otros tipos de transportadores existen en el interior del citoplasma y son las vesículas transportadoras de monoaminas.
En condiciones en las que una neurotoxina entra al citoplasma neuronal, tiende a ser secuestrada por compartimientos intracelulares, evitando la acción directa sobre las mitocondrías, situación que afectaría los sistemas respiratorios intracelulares.
Se ha encontrado una situación similar a la intoxicación con MPTP cuando se utilizan neurolépticos. En ambos casos se produce una inhibición de la actividad de las mitocondrias, dada por la afectación del complejo I de la cadena respiratoria. También se afectan, pero en menor grado los complejos II y III.
Cuando se estudia el fenómeno en mitocondrias que han sufrido fragmentación, las concentraciones requeridas de neurolépticos que inhiban el complejo I son más bajas que las concentraciones requeridas del metabolismo tóxico MPP.
En mitocondrías intactas, en cambio, las concentraciones requeridas de MPP no son demasiado diferentes de las concentraciones de neurolépticos requeridas para producir el mismo efecto.
Esto demuestra entonces que en las neuronas in vivo se necesitaría una alta concentración de haloperidol por ejemplo, y que no existe concentración del neuroléptico dentro de las mitocondrias.
Entonces, ¿cómo explicar la inhibición del sistema mitocondrial respiratorio?
Subramanyan y col. aciertan en clarificar el problema planteado cuando comparan los pasos metabólicos que siguen la toxina MPTP por un lado, y los neurolépticos como el haloperidol por otro.
En primer lugar, la estructura molecular del MPTP y del antipsicótico haloperidol muestran analogías:
- El MPTP se biotransforma en tetrahidro-piridium y en MPP o metil fenil-piridium (metabolito tóxico);
- El haloperidol se biotransforma sucesivamente por pasos de oxidación y deshidratación de su mitad heterocíclica, y deshidratación de su cadena piperidínica en tetrahidro-piridium haloperidol, y por alfa-carbono oxidación en haloperidol dihidro-piridium. Esta molécula a su vez se biotransforma por oxidación espontánea en haloperidol piridium.
Este último producto de biotransformación es similar biológicamente al MPP, y es que al igual que el metil fenil-piridium, ejercería un efecto tóxico sobre las neuronas dopaminérgicas, específicamente sobre las mitocondrias, afectando los procesos oxigenatorios o respiratorios celulares.
Los neurotransmisores se almacenan en vesículas secretorias que evitan lesiones intracitoplásmicas de sus organelos.
Al respecto, se ha demostrado la toxicidad de los neurotransmisores monoaminérgicos cuando éstos se vacían en el medio citoplasmático. Esto nos permite elaborar la hipótesis de que en la esquizofrenia se produciría un desequilibrio entre el transporte de la membrana plasmática y el transporte vesicular de monoaminas, lo que llevaría a un estado de acumulación de monoaminas en el citoplasma, con un efecto de daño celular que se va desarrollando en forma prolongada durante gran parte de la vida del paciente esquizofrénico.
El hecho de que las neuronas del tegmento ventral sean por ejemplo más resistentes a las injurias con MPTP que las células de la substancia negra, no indican de ninguna manera de que éstas sean invulnerables a un estado crónico de disrregulación vesicular y una disminución probable en el grado de clearence (depuración) de las toxinas endógenas o exógenas.
A los eventos propios del disturbio biológico, nuestra psicofarmacología neuroléptica o antipsicótica, utilizada hasta hace poco tiempo con criterios vagos (a mayores síntomas más dosis), conlleva el riesgo de producir productos tóxicos en la biotransformación que serían tomados por las neuronas dopaminérgicas y concentrados en la mitocondria, interfiriendo en la respiración celular por una selectiva inhibición de la NADH: ubiquinona oxirreductasa (en el complejo I), sucesos que terminarían con la vida celular.
Las concentraciones de clorpromazina y haloperidol capaces de inhibir el complejo I, son a nivel micromolar bajas, mientras que se requieren concentraciones altas del antipsicótico atípico clozapina para producir los mismos efectos.
3.3.5- Aminoácidos excitatorios y esquizofrenia
El glutamato y el aspartato son aminoácidos excitatorios que tienen correspondencia con los procesos fisiológicos de la consolidación de la memoria y en lo patológico con la activación de focos irritativos implicados en el disturbio epiléptico, o en los fenómenos del deterioro cognoscitivo de la enfermedad de Alzheimer, cuando las lesiones de las neuronas glutaminérgicas asientan en las vías aferentes intrínsecas y en las eferentes de la formación del hipocampo.
Existen tres tipos de receptores de los aminoácidos excitatorios:
- El NMDA (N-Metil-D-Aspartato);
- El AMPA (ácido Alfa Amino Hidroximetil-4-Isoxiasol), denominado también quiscualato;
- El kainato.
El receptor NMDA tiene una amplia distribución en las capas externas de la corteza cerebral, así como en el hipocampo y tálamo, entre otras estructuras cerebrales y medulares.
El receptor NMDA es un substrato donde actúan exotoxinas cerebrales, la PCP (fenciclidina) y otras psicomiméticas.
La conducta de estereotipias del esquizofrénico podría relacionarse con el control descendente antagónico que ejerce sobre el sistema dopaminérgico ascendente la activación de los receptores EAAS (aminoácidos excitatorios) localizados en la corteza.
4.- DROGAS RELACIONADAS CON LA DOPAMINA
Tres de las drogas más importantes relacionadas con la dopamina son: la cocaína, el éxtasis y las anfetaminas.
Cocaína
La cocaína es un alcaloide contenido en las hojas del arbusto «Erythroxylon coca» siendo químicamente un derivado de la latropina. Es un estimulante cerebral extremadamente potente, de efectos similares a las anfetaminas. Además, es un enérgico vasoconstrictor y anestésico local, siendo absorbido cuando se aspira nasalmente por las mucosas y metabolizado en el hígado, eliminándose por la orina. Fue utilizada inicialmente para el tratamiento de trastornos respiratorios y depresivos. Por sus efectos analgésicos, se utilizó en intervenciones quirúrgicas. Posteriormente se empleó con fines militares por su efecto vigorizante y el componente de agresividad que otorga. A comienzos del siglo XX se empezaba el consumo de cocaína por aspiración nasal. En esta época, eran prácticamente desconocidos sus efectos perjudiciales, por lo que estaba presente en las fórmulas de bebidas, jarabe contra la tos, lociones capilares, y hasta cigarrillos. En 1909 existían en EE.UU. más de 70 bebidas registradas con componentes de cocaína, lo que incrementó la producción en los países donde se cultivaba coca, fundamentalmente Perú. Los estudios del uso de cocaína comenzaron, con FREUD, al que siguieron HEMMOND (1887) y BOSE (1902), los cuales encontraron sintomatología aguda y crónica en el consumo. Más recientemente, a partir de la década de los ochenta, los experimentos sobre patrones de consumo y cantidades constataron sus efectos sobre la adrenalina, muy relacionada con la agresividad.
La vigabatrina incrementa los niveles de un neurotransmisor conocido como GABA. Los altos niveles de GABA producen bajos niveles de otro neurotransmisor, la llamada dopamina. Este compuesto es el responsable de la sensación de bienestar que forma la base del efecto de las drogas.
Y al reducir los niveles de dopamina en el cerebro, los niveles de ansiedad de los adictos también bajan.
Mediante estudios del cerebro de los simios, los investigadores determinaron que los animales tratados con vigabatrina mostraban niveles normales de dopamina incluso después de consumir una dosis de cocaína, mientras que el resto presentaban niveles altos.
En otro experimento, los científicos administraron cocaína a ratas hasta lograr una adicción, y observaron que los animales tendían a volver una y otra vez al sitio donde habían conseguido la droga. Pero una vez que fueron tratados, los roedores no se quedaban en el lugar asociado con la cocaína sino que se movían normalmente por toda la jaula.
Dewey dijo que el hallazgo es importante para las personas adictas a la cocaína y otras drogas, porque su ansiedad puede ser activada por factores muy diversos, como ver sustancias de aspecto parecido a la droga que consumían o a una persona con la que compartían el hábito.
"Como la adicción a la cocaína tiene componentes bioquímicos y de comportamento, estos resultados confirman que es posible atacarla en ambos frentes", dijo Charles Ashby, un investigador de la Universidad de St. John's que trabajó en los aspectos psicológicos del estudio.
La cocaína estimula el sistema nervioso central, actuando directamente sobre el cerebro. Sus efectos fisiológicos son inmediatos; entre ellos pueden citarse: sudoración, aumento en la potencia muscular, midriasis (dilatación de las pupilas), incremento de actividad cardíaca y presión sanguínea, dilatación de los vasos sanguíneos periféricos, convulsiones, aumento en el ritmo respiratorio y de la temperatura corporal. Estos síntomas pueden provocar la muerte por paro cardíaco o fallas respiratorias. Además se presentan irritaciones y úlceras en la mucosa nasal. Comúnmente causa congestión nasal, que puede presentarse o no con secreción liquida. El uso por vía inyectable, expone al adicto a infecciones de Sida, hepatitis y otras enfermedades infectocontagiosas.
La cocaína es una sustancia extremadamente adictiva, cuyos efectos se perciben en un lapso de 10 segundos y duran alrededor de 20 minutos. Actúa directamente sobre los centros cerebrales encargados de las sensaciones del placer. Dada su capacidad extremadamente alta de producir daños y hasta destrucción celular, las sensaciones que eran placenteras en sujetos recién iniciados, se convierten en efectos desagradables como agitación, llanto, irritabilidad, alucinaciones visuales auditivas y táctiles, delirio paranoide, amnesia, confusión, fobias o terror desmedido, ansiedad, estupor, depresión grave y tendencias suicidas.
Los efectos psíquicos reconocidos por la mayoría de los autores y recogidos en publicaciones recientes incluyen euforia, inestabilidad, aumento de la comunicación verbal, y de la seguridad en uno mismo, inquietud, anorexia, insomnio e hipomanía. El adicto experimenta pérdida de interés e imposibilidad de sentir placer ante la falta de la sustancia. Así, la cocaína se convierte en el único objetivo y motivo en la vida del adicto, desplazando todo tipo de sentimientos.
La relación con los fenómenos criminales son expresamente citados por los autores, asociándose su consumo a la predisposición al delito. La cocaína es consumida por muy variados tipos de sujetos y motivos. Existe un patrón de consumo recreativo, al estilo del consumo del alcohol, presentando una ingesta controlada de la sustancia; es el caso de quienes ingieren la droga ocasionalmente cuando se les ofrece. Se diferencian radicalmente de adictos habituales, quienes desarrollan tolerancia y necesitan de mayores dosis para alcanzar iguales resultados. A esta situación puede llegarse por causas diversas pero siempre relacionadas con factores sociales y ambientales determinantes.
La adicción a la cocaína posee unos condicionantes desencadenantes, que pueden ser el reforzamiento de una personalidad insegura, que recibe un apoyo en el estímulo del tóxico. En lugar de tratar este déficit patológico con antidepresivos o fármacos estabilizadores del estado de ánimo se recurre a una vía aparentemente rápida. Dado que los efectos de la cocaína sobrepasan su punto álgido a los treinta minutos, el individuo precisa varias dosis durante el día para alcanzar cierta estabilidad emocional y evitar el efecto disfórico que la propia droga ocasiona cuando transcurren varias horas desde la ingesta.
El uso de cocaína es altamente susceptible de producir daños irreparables en recién nacidos, cuyas madres mantuvieron su adicción durante el embarazo (esto último hizo que algunos Estados de los Estados Unidos de América sometan a las adictas embarazadas a prisión, mientras dura el embarazo).
Éxtasis o MDMA
La metilendioximetanfetamina (MDMA), normalmente conocida como "éxtasis", "ectasi" o "X-TC", es una droga sintética sicoactiva con propiedades alucinógenas de gran potencial emotivo y perturbador psicológico, con propiedades similares a las anfetaminas. Su estructura química (3-4 metilendioximetanfetamina) se asemeja a la estructura de la metilendioxianfetamina (MDA) y de la metanfetamina, otros tipos de drogas sintéticas causantes de daños cerebrales. Durante los años sesenta se utilizó con fines terapéuticos dado que según determinados sectores de la psiquiatría ayudaba a la comunicación y al tratamiento de neurosis fóbicas. Surgió entonces la polémica médico - legal, atribuyendo a su consumo repercusiones en la delincuencia, por lo que finalmente fue ilegalizado.
El éxtasis produce efectos psíquicos de gran potencial perturbador. Inicialmente el sujeto experimenta sensaciones de confianza y excitación, a las que sigue un estado de hiperactividad e incremento en los pensamientos morbosos. Los efectos del estimulante se diluyen provocando trastornos psicológicos, como confusión, problemas con el sueño (pesadillas, insomnio), deseo incontenible de consumir nuevamente drogas, depresión, ansiedad grave y paranoia. Estos efectos han sido reportados incluso luego de varias semanas del consumo. También se han informado casos graves de psicosis. Entre los síntomas físicos pueden citarse: anorexia, tensión y trastornos musculares similares a los presentes en la enfermedad de Parkinson, bruxismo (apretamiento involuntario de los dientes), náuseas, visión borrosa, nistagmus (movimientos oculares rápidos), desmayos, escalofríos y sudoración excesiva (este último signo es característico durante la intoxicación). El aumento de la frecuencia cardíaca y la tensión arterial, crea riesgos de trastornos circulatorios o cardíacos. Informes forenses indican que en personas con definiencias cardiorespiratorias puede producir muerte súbita.
La MDA, el fármaco de origen de la MDMA, es una droga similar a la anfetamina que también ha sido abusada, presentando efectos psico-físicos similares a los de la MDMA. Las investigaciones han mostrado que la MDA destruye las neuronas productoras de serotonina, que regulan directamente la agresión, el estado de ánimo, la actividad sexual, el sueño y la sensibilidad al dolor. Es probable que esta acción sobre el sistema productor de serotonina sea el origen de las propiedades síquicas.
La MDMA también guarda relación en su estructura y sus efectos con la metanfetamina, la cual ha demostrado ser causante de la degeneración de las neuronas que contienen la sustancia neurotransmisora dopamina.
En experimentos de laboratorio, una sola exposición a la metanfetamina en dosis elevadas o el uso prolongado en dosis bajas destruye hasta un 50% de las células cerebrales. Aunque este daño tal vez no sea aparente de inmediato, los científicos creen que con el envejecimiento o la exposición a otros agentes tóxicos, pueden aparecer síntomas de la enfermedad de Parkinson con el tiempo. Estos comienzan con falta de coordinación y temblores y a la larga pueden causar una forma de parálisis.
Anfetaminas
Se trata de otras drogas que también reducen los niveles de dopamina en el organismo, al igual que lo hace la cocaína, no obstante sus efectos son más duraderos y profundos.
Fueron sintetizadas por primera vez entre la última década del siglo XIX y la primera del siglo XX. Los primeros experimentos clínicos se iniciaron hacia 1930, y desde 1935 se comercializó con gran difusión en el Reino Unido, Francia y Alemania. Durante la Segunda Guerra Mundial fue utilizada indiscriminadamente por todos los bandos, dado el carácter euforizante que contiene la sustancia y la agresividad otorga.
El consumo de este excitante está ampliamente extendido y distribuido por todas las clases sociales. A diferencia de lo que sucede con la cocaína que la consumen preferentemente los sectores medios y altos, las anfetaminas son consumidas tanto por ejecutivos que pretenden sobreexcitación como por amas de casa que buscan un anoréxico para sus dietas o por estudiantes que preparan exámenes. Al incidir en el sistema ortosimpático causan hipertensión, taquicardia, hiperglucemia, midriasis, vasodilatación periférica, hiperpnea, hiporexia, etc. El estado de ánimo del adicto oscila entre la distrofia y la hipomanía, así como ansiedad, insomnio, cefalea, temblores y vértigo. Pueden aparecer cuadros depresivos y síndromes paranoides anfetamínicos. A dosis normales, sus efectos varían de acuerdo al individuo y las condiciones de ingesta. Pueden producir efectos placenteros, hiperactividad y sensación desbordante de energía, pero también causan temblor, ansiedad irritabilidad, ira inmotivada y repentina y trastornos amnésicos e incoherentes. En la última fase se describen depresión, cuadros paranoides y delirios paranoides, alucinaciones y trastornos de conducta. El consumo de anfetaminas puede conducir a actuaciones agresivas, al igual que los barbitúricos y el alcohol, por su gran efecto euforizante, unido a un descontrol en los instintos inhibitorios. Tales situaciones se producen cuando las dosis suministradas, generalmente por vía endovenosa, superan los dos gr. Está demostrado un mayor potencial en las anfetaminas que en la cocaína, tanto en su punto más álgido como en la duración de los efectos. Reacciones muy graves se producen al consumirlas con barbitúricos en el conocido fenómeno de la pluritoxicomanía. Tomadas en dosis importantes son causantes de confusión, tensión, ansiedad aguda y miedo. También pueden precipitar psicosis paranoide en sujetos no psicóticos. La psicosis anfetamínica desarrollada por el sujeto se asemeja a la psicosis paranoica y a la esquizofrenia paranoica.
1
2
Descargar
| Enviado por: | Doraimon |
| Idioma: | castellano |
| País: | España |
Todos los derechos reservados.