Biología
ADN (Ácido Desoxirribonucleico), Hibridación y reacción en cadena de las Polimerasas
I. Estructura y características básicas del material genético
Los ácidos nucleicos fueron descubiertos en 1869 por Miescher, que consiguió precipitar una sustancia rica en fósforo, que denominó nucleína ya que procedía de los núcleos de las células, a partir del pus recogido de vendas en un hospital. Esta sustancia fue posteriormente denominada ácido nucleico y más tarde ácido desoxirribonucleico (DNA).
En la década de 1920 se elucidó la composición química del DNA, estableciéndose que contenía cuatro bases nitrogenadas: dos bases púricas (adenina y guanina) y dos pirimidínicas (timina y citosina), un azúcar (desoxirribosa) y ácido fosfórico. La unión de una base nitrogenada y la desoxirribosa origina un nucleósido, mientras que los nucleótidos contienen un nucleósido y una o más moléculas de ácido fosfórico (Figura 1.1). Si en la composición del ácido nucleico el azúcar es ribosa y en lugar de timina hay uracilo (otra base pirimidínica), se habla de ácido ribonucleico (RNA).
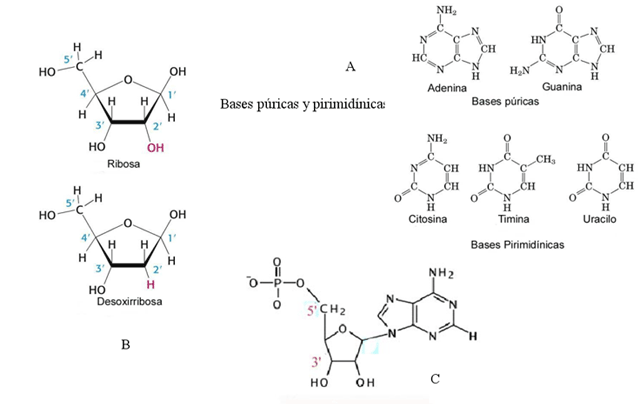
Figura 1.1
1A, Estructura de las bases púricas y pirimidínicas; 1B, los azúcares ribosa y desoxirribosa; 1C, nucleótido monofosfato de adenosina (nótese la numeración 5'-3')
En esta primera mitad del siglo XX se sucedieron los descubrimientos relacionados con los ácidos nucleicos, demostrándose que eran los portadores de la información genética.
En 1953 Watson y Crick publican su conocido trabajo en Nature (http://www.nature.com/nature/dna50/watsoncrick.pdf) en el que proponen la estructura de doble hélice para el DNA, con dos hebras antiparalelas (Figura 1.2), apareándose la adenina con la timina mediante dos enlaces de puentes de hidrógeno y la guanina con la citosina mediante tres enlaces de puentes de hidrógeno (Figura 1.3); esto implica que las dos hebras de la hélice son complementarias en cuanto a su composición en bases nitrogenadas.
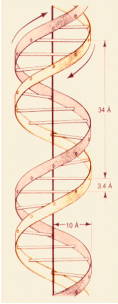
Figura 1.2
Estructura de la doble hélice de DNA, mostrando la orientación de cada hebra. El eje interno tiene un radio de 10Å; cada giro de la hélice recorre 34Å, que suponen 10 bases nitrogenadas (el espacio entre cada base es de 3,4Å)
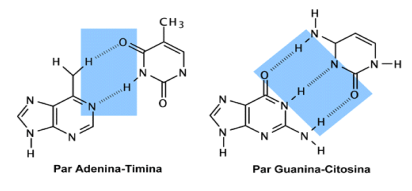
Figura 1.3
Enlaces entre los pares de bases A-T y G-C
El ácido ribonucleico (RNA) sin embargo, además de las diferencias en composición ya comentadas, suele tener una única cadena, que puede enrollarse sobre sí misma para formar estructuras en forma de horquilla.
El DNA es el portador de la información genética. El flujo de la información genética va del DNA al RNA y finalmente a las proteínas, aunque hay algunas excepciones a esta norma (algunos virus RNA, por ejemplo). La información del DNA está contenida en genes, que son entidades físicas que dan lugar a un polipéptido. Esta información se transcribe a RNA mensajero (RNAm), que es una copia complementaria del gen. La secuencia de nucleótidos del RNAm se transforma en secuencia de aminoácidos en el proceso de traducción, en el que también interviene otro tipo de RNA, denominado de transferencia (RNAt). Un tercer tipo de RNA (RNA ribosómico, RNAr) también interviene en la traducción, como integrante de la estructura de los ribosomas. Los RNAt y RNAr también están codificados en el DNA.
El código genético fue descifrado en la década de 1960 por varios investigadores (Khorana y Nirenberg entre otros; en este apartado fue esencial la aportación de Severo Ochoa). Cada aminoácido está codificado por un grupo de tres bases (triplete), si bien este código está degenerado, ya que la mayor parte de los aminoácidos están codificados por más de un triplete.
El DNA se replica mediante la acción de DNA polimerasas, enzimas que catalizan la adición de nucleótidos a una estructura molecular inicial. La replicación comienza en un punto, en el que se abre la doble hélice y, en el proceso de réplica, se copian las dos cadenas. Las DNA polimerasas van añadiendo nucleótidos en dirección 5'-3' (la dirección se indica de acuerdo a la numeración de los átomos de carbono del azúcar, como se observa en la figura 1.1 C), pero para comenzar necesitan que exista un grupo hidroxilo libre en 3'; por ello es necesario que exista un iniciador. Este proceso de replicación es semiconservativo, ya que en cada nueva doble hélice hay una hebra de nueva síntesis y otra vieja, procedente del DNA que se copia (Figura 1.4). Además de poseer actividad polimerasa, las DNA polimerasas también presentan actividad exonucleasa 3'-5', que permite comprobar y corregir los errores que se produzcan durante la replicación.
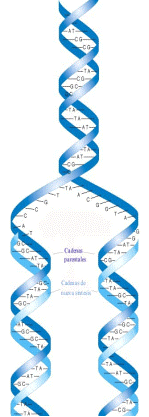
Figura 1.4
Replicación de una molécula de DNA para dar lugar a 2 nuevas moléculas. La mitad de las cadenas proceden de la molécula original
Aunque los DNAs de todos los organismos son similares en estructura y composición, en las células eucariotas el DNA se localiza en los cromosomas, pero en las células procariotas (bacterias) se encuentra como un único círculo situado en una región denominada nucleoide, aunque es común denominarlo cromosoma bacteriano. Además de este nucleoide, existen otros DNAs bacterianos, como son los plásmidos, pequeñas moléculas circulares de DNA que existen de forma independiente. Estos plásmidos contienen genes que pueden ser importantes para las bacterias. Entre los distintos tipos de plásmidos existentes, destacan los plásmidos R, que contienen genes que confieren resistencia a antibióticos, y los plásmidos de virulencia, que incrementan la virulencia de algunas bacterias al conferirles capacidad de producir toxinas o de resistir a las defensas de la célula huésped. Algunos plásmidos son conjugativos, es decir que son capaces de transferirse de una célula a otra. Además, en este proceso de conjugación, también se pueden movilizar genes cromosómicos.
Dentro del cromosoma de la bacteria (y también de las células eucariotas) hay fragmentos de DNA que tienen la capacidad de variar su posición en el genoma, en el fenómeno conocido como transposición. Hay varios tipos de elementos transponibles: secuencias de inserción, transposones, algunos tipos especiales de virus. Sus características fundamentales son la presencia de secuencias terminales repetidas e invertidas (por ejemplo: ACCG- secuencia de inserción -CGGT) y la presencia de genes que codifican una transposasa, enzima implicada en la transposición.
El avance en el conocimiento de los plásmidos permitió en 1973 a Cohen y Boyer construir el primer plásmido recombinante (con genes procedentes de distintos orígenes) capaz de replicarse en un huésped bacteriano, dando lugar al nacimiento de la ingeniería genética, mediante la cual es posible la transferencia artificial de información genética entre organismos.
II. Fundamento de las técnicas genéticas aplicadas a la identificación y detección de bacterias y virus en alimentos y aguas
II.1. Hibridación
II.1.1. El proceso de hibridación
Desde los primeros estudios con DNA, se supo que se podían cambiar sus propiedades físicas mediante el empleo de calor o modificando el pH. Durante el calentamiento de una solución de DNA, cuando la temperatura alcanza el punto de fusión del DNA (Tm, melting point, es la temperatura a la que la mitad del DNA presente está desnaturalizado; esta temperatura varía para los DNAs de los diferentes organismos, dependiendo sobre todo del contenido en pares guanina-citosina, G-C, que confieren mayor estabilidad), las hebras se separan produciéndose la desnaturalización. Este proceso es reversible y las hebras individuales de DNA pueden volver a asociarse cuando se restablecen las condiciones de temperatura (Figura 1.5A). Si en una solución tenemos DNAs de distintas procedencias que tengan regiones complementarias, también se pueden reasociar las distintas hebras dando lugar a moléculas híbridas (hibridación; Figura 1.5B).
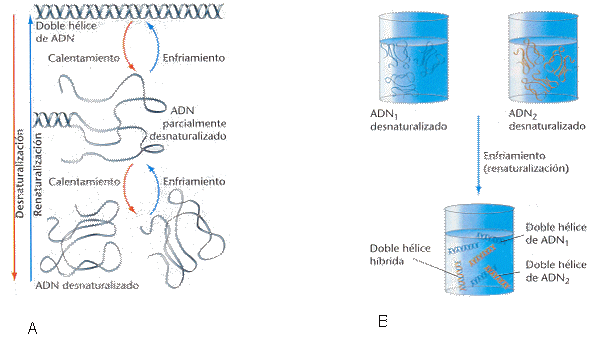
Figura 1.5
5A, Desnaturalización y renaturalización por calentamiento/enfriamiento
de una molécula de DNA
5B, Mezcla de dos soluciones conteniendo DNA desnaturalizado
para dar lugar a moléculas híbridas de DNA
II.1.2. Sondas de hibridación
Con los conocimientos que existen de los genomas de los organismos, cada vez es más sencillo generar pequeños fragmentos de DNA (oligonucleótidos) que sean complementarios de regiones que nos interesan, por ejemplo de genes específicos de una especie bacteriana. De esta forma, si en una muestra problema se produce la hibridación entre el oligonucleótido y el DNA presente en la muestra, se puede concluir que la bacteria de interés estaba presente en esa muestra. A estos fragmentos de DNA se les denomina sondas.
II.1.3. Factores de los que depende el proceso de hibridación
Los factores que más influyen en el proceso de hibridación son la temperatura, el pH y la concentración de cationes. La modificación de estos parámetros condiciona la fidelidad con la que se produce la hibridación (en inglés se denomina stringency, traducido en ocasiones como "“astringencia”"), es decir, el número de pares de bases erróneos que puede tolerar un híbrido. Si se emplean soluciones con baja concentración salina o la reacción se lleva a cabo a temperatura elevada, es poco probable que se forme un híbrido estable (hibridación de gran fidelidad o de "high stringency"). Estas condiciones se usan para evitar la aparición de falsos resultados positivos.
Por el contrario, las soluciones con alta concentración salina o las bajas temperaturas permiten la formación de híbridos imperfectos (algunos pares de bases no están correctamente emparejados). Estas condiciones de baja fidelidad (“low stringency”) se usan para detectar secuencias relacionadas con la de la sonda, aunque no sean perfectamente complementarias (por ejemplo, para géneros de bacterias o para detectar genes relacionados con algún gen ya caracterizado).
II.1.4. Detección de la hibridación (i): marcadores
Para poder observar si la sonda es híbrida o no con el DNA problema, es necesario "marcar" la sonda de forma que pueda dar una señal medible. Los primeros marcadores empleados eran isótopos radiactivos, normalmente P32. De esta forma, se podían detectar los híbridos mediante la exposición de películas fotográficas; sin embargo, las sondas marcadas con P32 tienen poca vida útil, ya que este isótopo tiene una vida media de 14 días, y además obliga al manejo de material radiactivo.
Actualmente existen diversas alternativas no radiactivas para marcar las sondas, como puede ser el uso de enzimas (peroxidasa o fosfatasa, que llevan a cabo una reacción detectable), marcadores de afinidad (biotina o digoxigenina, que se van a unir posteriormente a otra molécula) o moléculas quimioluminiscentes (que producen luz y puede excitar una película fotográfica).
La molécula marcadora se puede incorporar a la sonda mediante distintas técnicas:
• Unión al azar ("random priming"). Consiste en copiar un fragmento de DNA utilizando una DNA polimerasa y añadiendo a la reacción algunos nucleótidos marcados; se incorpora un nucleótido marcado cada 20-25 bases.
• Marcado por relleno ("nick translation"). Se realizan cortes enzimáticos ("nicks") en el DNA y después se reparan con una DNA polimerasa utilizando nucleótidos marcados.
• Marcado en 3'. Se añade un único nucleótido marcado en el extremo 3' empleando una transferasa terminal.
• Marcado extendido en 3' ("3' tailing"). Similar a la técnica anterior, pero se añade una cadena sencilla de 10-50 bases en el extremo 3'.
• Marcado en 5'. Se pueden marcar las sondas en el extremo 5' durante su síntesis química.
• Marcado de RNA. Se usan una RNA polimerasa y nucleótidos marcados para que se transcriba un fragmento de DNA, dando lugar a un RNA marcado.
• Marcado por PCR. Se lleva a cabo una PCR (ver más adelante) empleando nucleótidos marcados para amplificar un fragmento de DNA.
• Marcado por fotoactivación. Se une un marcador covalentemente al DNA mediante exposición a la luz UV.
La mayor parte de estas técnicas de marcado están disponibles en forma de “kits” comerciales y la elección de alguna de ellas depende del uso al que se vaya a destinar la sonda y de la cantidad de marcador que se quiera incorporar (por ejemplo, una sonda marcada en 3' será más específica pero menos sensible que la misma sonda marcada en 3' extendido, ya que la primera tiene menos marcador incorporado, pero la cadena sencilla añadida a la segunda puede permitir que hibride con menor especificidad).
II.1.5. Detección de la hibridación (ii): formatos de realización de la hibridación
Hay diversos formatos de utilización de las técnicas de hibridación:
- Formato líquido-líquido. Tanto el DNA problema como la sonda marcada están en solución. Esto permite una reacción más rápida, pero es más difícil la detección, ya que, habitualmente, hay que separar la sonda que ha hibridado de la que no ha hibridado (frecuentemente por diferencias en solubilidad). Por esta razón, este formato no es muy utilizado en los sistemas comerciales, aunque se está empleando cada vez con más frecuencia en las aplicaciones de PCR en tiempo real (ver más adelante).
- Formato líquido-sólido: En este formato uno de los componentes de la reacción (el DNA problema o una sonda) está en solución, mientras que el otro está unido a un soporte sólido. En los laboratorios de investigación se suele fijar a un filtro el DNA, que luego se desnaturaliza e hibrida con la sonda (hibridación "dot-blot" o "slot-blot"; Figura 1.6). Sin embargo, en algunos sistemas comerciales (por ejemplo, Gene-Trak, http://www.neogen.com/genetrakback.htm, o LumiProbe) inmovilizan una sonda (de captura) a un soporte y el DNA problema se añade en solución; finalmente, una segunda sonda (marcada) permite detectar la hibridación.
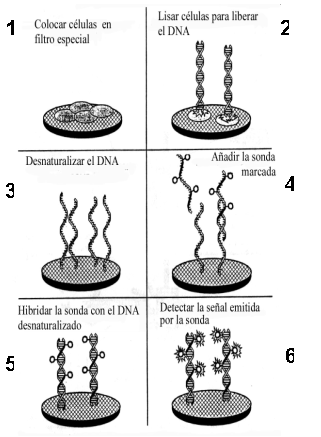
Figura 1.6
Etapas en la hibridación con una sonda marcada de muestras de DNA, preparadas a partir de células fijadas a un filtro
- Hibridación in situ. Este modelo permite detectar el DNA problema en el interior de las células, permeabilizándolas de alguna forma que permita la entrada de la sonda. Cuando la sonda está marcada con una molécula que emite fluorescencia se habla de FISH (fluorescence in situ hybridization; Figura 1.7).
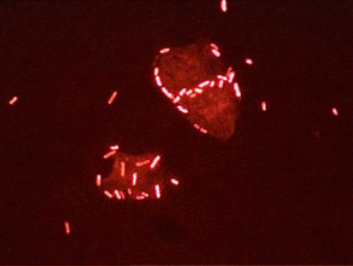
Figura 1.7
FISH (fluorescent in situ hybridization); las células se visualizan debido a que su DNA ha hibridado con una sonda marcada con una molécula fluorescente
Aunque en el momento presente las técnicas de hibridación no están muy extendidas para el análisis rutinario de bacterias de interés en alimentos, hay que tener presente que los sistemas más prometedores para la detección rápida, la PCR en tiempo real y los “chips” (“arrays”) de DNA se basan en gran medida en el uso de sondas específicas.
II.2. Técnicas de amplificación de ácidos nucleicos: La reacción en cadena de la polimerasa (PCR) y sus variantes
II.2.1. Descripción de la reacción en cadena de la polimerasa
En 1983, Mullis desarrolló un procedimiento que permitía realizar copias de un fragmento de DNA de forma rápida y económica y que se denominó reacción en cadena de la polimerasa (PCR). Esta técnica supuso un gran avance para la universalización de las técnicas moleculares en los laboratorios de microbiología, por su sencillez y facilidad de aplicación.
La PCR se basa en la repetición de tres procesos: (i) desnaturalización térmica del DNA; (ii) hibridación de cebadores (oligonucleótidos) específicos al DNA de cadena sencilla, y (iii) extensión enzimática del DNA (Figura 1.8). El proceso se lleva a cabo en un bloque térmico denominado termociclador. En cada repetición se duplica la cantidad de DNA específico, así en una reacción típica de 30-40 ciclos se generan 230-40 moléculas del DNA de interés (amplicones).
La temperatura de hibridación de los cebadores condiciona la especificidad del proceso; cuando no se conoce perfectamente la secuencia del DNA de interés se puede utilizar una temperatura inferior a la óptima o también se pueden diseñar cebadores degenerados, en los que alguna de las posiciones puede estar ocupada por más de un nucleótido.
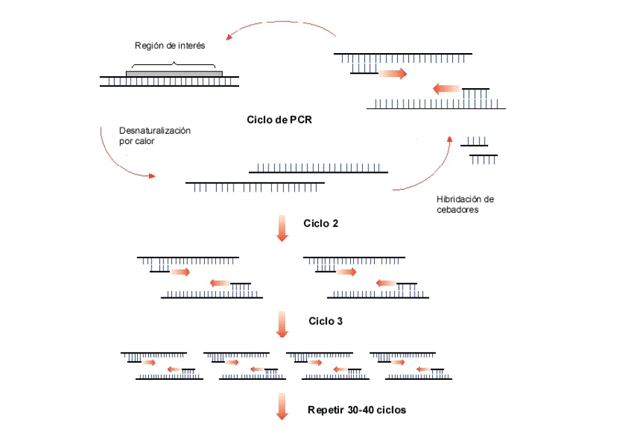
Figura 1.8
Esquema de la reacción en cadena de la polimerasa (PCR), mostrando 3 ciclos de amplificación; al finalizar el tercer ciclo se han generado 8 moléculas de DNA (23)
II.2.2. Materiales necesarios para la realización de una PCR y precauciones básicas
En el tubo de reacción hay que añadir un tampón de reacción adecuado, con una concentración de Mg2+ óptima para el funcionamiento enzimático, los cebadores específicos (aprox. 0,4 µM de cada uno), los desoxinucleótidos trifosfato (dATP, dTTP, dGTP y dCTP; 200 µM de cada uno), la DNA polimerasa (1 U) y la muestra de DNA (entre 50-250 ng). El uso de DNA polimerasas termoestables permite que la enzima no se desnaturalice en la etapa de calentamiento.
Debido a la elevada sensibilidad de la PCR es esencial evitar contaminaciones por DNA extraño; para ello se separan físicamente las zonas de preparación de muestras, preparación de reactivos, amplificación y detección; además, es conveniente el uso de puntas de pipeta con filtros, guantes desechables y pipetas diferenciadas para manipular muestras con DNA y reactivos. Es obligado el uso de controles positivos y negativos en cada tanda de reacciones.
II.2.3. Lectura de los resultados de la PCR
En la PCR convencional, la visualización de los resultados se realiza al final del proceso. La forma más habitual conlleva la separación electroforética de los fragmentos amplificados en un gel de agarosa, la posterior tinción con bromuro de etidio (u otro colorante fluorescente) y la observación final transiluminando con luz ultravioleta. Se utilizan patrones de peso molecular conocido que permiten extrapolar el tamaño (pares de bases, pb) de los fragmentos amplificados (Figura 1.9). Otras posibilidades de detección de los fragmentos pasan por el uso de sondas marcadas, como ya se comentó para la hibridación, que se unan específicamente al amplicón.
II.2.4. Variantes de la PCR
Existen numerosas variantes de la técnica original de PCR, algunas de las cuales se comentan a continuación:
· PCR mútiple (multiplex PCR): Se pueden incluir en la misma reacción diversos pares de cebadores que amplifiquen fragmentos diferentes de DNA; de esta forma se pueden detectar genes diversos de un mismo organismo o detectar diferentes organismos al mismo tiempo (Figura 1.10). Los cebadores deben de tener una estabilidad térmica similar y puede ser necesario modificar algunas condiciones de la reacción para evitar interacciones entre los cebadores.
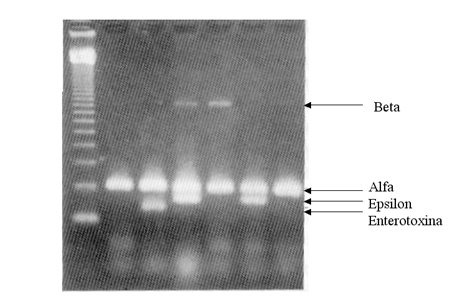
Figura 1.10
Gel en el que se muestra el resultado de una PCR múltiple para detectar los genes que codifican para las toxinas á, â, å y la enterotoxina de Clostridium perfringens
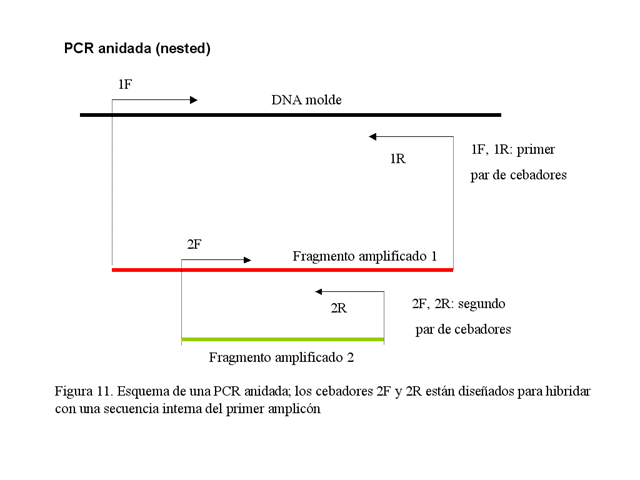
· PCR anidada (nested PCR): En esta modalidad, se amplifica un fragmento y a continuación se usa este amplicón como molde para una segunda reacción empleando cebadores que hibriden con una secuencia interna del primer fragmento (Figura 1.11). De esta forma se aumentan la sensibilidad y la especificidad de la reacción, ya que se pueden usar condiciones menos específicas en la primera amplificación y más específicas en la segunda.
· PCR con retrotranscripción (Reverse Transcription-PCR, RT-PCR): Se utiliza cuando el molde es RNA, que primero se copia a DNAc (DNA complementario al RNA) mediante una transcriptasa inversa. En microbiología alimentaria, por ejemplo, se usa para la detección de aquellos virus cuyo material genético está constituido por RNA (véase el capítulo correspondiente de este mismo curso).
II.2.5. Otras técnicas de amplificación de ácidos nucleicos
Además de la PCR, otras técnicas de amplificación de ácidos nucleicos tienen interés diagnóstico. Las de mayor importancia son:
• Reacciones isotérmicas: Existen algunas modalidades de amplificación que permiten trabajar a la misma temperatura durante todo el proceso, eliminando la necesidad de contar con un termociclador:
- En la amplificación por desplazamiento de hebra (Strand Displacement Amplification, SDA) se van sintetizando cadenas sencillas de DNA a partir del molde original y estas cadenas sencillas sirven de molde para nuevas copias.
- En la amplificación basada en la transcripción (Transcription-based Amplification System, TAS; Self-sustained Sequence Replication, 3SR) el ácido nucleico de partida es RNA (que se puede generar a partir de DNA); así pues, la primera etapa consiste en copiar el DNAc a RNA para, a continuación, añadir RNA polimerasa a fin de sintetizar múltiples copias de RNA. Con sólo 4 ciclos de reacción se pueden generar más de 1 millón de copias de RNA.
• Hay otras modalidades de amplificación que utilizan sondas de hibridación y éstas son a su vez amplificadas, detectándose el producto de esta segunda reacción (reacción en cadena de la ligasa, LCR; replicasa QB).
• Otro tipo de técnicas son las que incrementan la sensibilidad de la detección amplificando la señal. Para ello se usan sondas de hibridación que por un extremo hibridan con el DNA de interés y por el otro están ramificadas y sirven para la unión de varias sondas marcadas, lo que confiere más intensidad a la señal emitida.
II.3. PCR en tiempo real (real time PCR) o PCR cuantitativa
II.3.1. Justificación de la necesidad de esta técnica
Aunque la utilización de la PCR en los laboratorios de microbiología alimentaria está ya bastante extendida, los protocolos de PCR convencional presentan algunas desventajas, como:
• La detección se produce al final de la reacción.
• La visualización se lleva a cabo en gel de agarosa tras su tinción con bromuro de etidio (un proceso lento y que implica el uso de un reactivo tóxico).
• Los resultados se basan en el tamaño de banda.
• La cuantificación se basa en la intensidad de tinción y es imprecisa.
II.3.2. Fundamento de las técnicas de PCR en tiempo real
Una modificación de las técnicas de amplificación que puede solucionar algunos de estos inconvenientes es la PCR en tiempo real, también denominada PCR cuantitativa. Se basa en detectar la fluorescencia emitida cuando se genera el fragmento específico durante la amplificación. Existen diversos formatos de detección que, en teoría, se pueden utilizar con todos los equipos de PCR en tiempo real, aunque cada fabricante recomienda el formato más adecuado para su(s) sistema(s).
II.3.3. Alternativas para el seguimiento del proceso de amplificación: formatos de realización de la PCR en tiempo real
El formato más sencillo es el que usa moléculas fluorescentes que se intercalan en el DNA de cadena doble, siendo la más utilizada SYBR® Green (menos tóxica que el bromuro de etidio; (Figura 1.12).

Figura 1.12
Esquema del mecanismo de acción de SYBR Green
El uso de intercaladores fluorescentes, sin embargo, presenta algunos inconvenientes, como que cualquier producto amplificado (específico o inespecífico) producirá fluorescencia o que no se puede utilizar para detectar productos múltiples, aunque hay técnicas para solventarlos.
El resto de formatos de detección se basan en la hibridación de sondas internas con el fragmento amplificado, existiendo diversos sistemas de generación de la señal fluorescente. Dentro de estos sistemas, los más conocidos son los siguientes:
· Sondas Taqman®. Diseñadas por Applied Biosystems, hacen uso de la actividad 5' exonucleasa de la Taq (Thermus aquaticus) DNA polimerasa. Estas sondas tienen un marcador fluorescente en el extremo 5' y una molécula que absorbe ("quencher") la fluorescencia emitida por el marcador en el otro extremo. La sonda hibrida con el fragmento específico (si está presente) y, a medida que se sintetiza la hebra complementaria al fragmento, la sonda se va degradando por la acción exonucleasa, liberando el marcador que ahora sí emite fluorescencia (Figura 1.13).

Figura 1.13
Principio de acción de las sondas Taqman
• Cebadores fluorescentes ("Molecular Beacons"). En estos tipos, la molécula fluorescente y la molécula que absorbe la fluorescencia se mantienen próximas mediante la formación de una horquilla de hibridación. Esta horquilla se abre durante la amplificación, aumentando la emisión de fluorescencia, como se muestra en la figura 1.14.


Figura 1.14: Esquema del funcionamiento de una sonda “molecular beacon”
• Doble sonda marcada o sondas de hibridación (sondas "LightCycler"). En este caso se emplean dos sondas marcadas con moléculas fluorescentes en posición 5' (sonda A) y 3' (sonda B) y que hibridan en regiones adyacentes del DNA diana. Cuando se produce la hibridación, la primera sonda emite fluorescencia que excita al colorante de la segunda sonda, produciendo una señal detectable (Figura 1.15).

Figura 1.15
Principio de funcionamiento de las dobles sondas marcadas; la fluorescencia emitida por la sonda A excita al fluorocromo de la sonda B, que a su vez emite fluorescencia
II.3.6. Lectura e interpretación de los resultados de una PCR en tiempo real
La visualización de los resultados se consigue mediante la integración de la señal fluorescente y se refleja en una curva de amplificación, considerándose que hay un resultado positivo cuando la señal emitida es superior al umbral establecido; el ciclo en que se considera que se obtiene la señal positiva está relacionado con la cantidad inicial de DNA molde (Figura 1.16).

Figura 1.16
Ejemplo de gráfica obtenida en un experimento de PCR en tiempo real para la detección de Listeria monocytogenes en alimentos inoculados con los niveles que se indican
Descargar
| Enviado por: | Maranata |
| Idioma: | castellano |
| País: | España |
Todos los derechos reservados.