Biología, Botánica, Genética y Zoología
Síndrome de Turner
1.- DEFINICIÓN Y TERMINOLOGÍA
Fue descrito por primera vez en 1930 por el Dr. Ullrich en Alemania. Alcanzó su divulgación en EEUU gracias a Henry Turner en 1938, con la presentación de varios casos de pacientes afectados. Ford y Cols fueron quienes, en 1959, reconocieron la base cromosómica del síntoma presentando a pacientes dotados con 45 cromosomas con un único cromosoma X.
Podríamos definir el síndrome como un trastorno con pérdida total o parcial de un cromosoma X o una anomalía de su estructura.
2.- EPIDEMIOLOGÍA DEL SÍNDROME
Este síndrome se presenta generalmente en mujeres, rara vez en hombres.
El síndrome de Turner es uno de los trastornos cromosómicos más frecuentes, afecta a una niña por cada 2.500 recién nacidas vivas, o lo que es lo mismo una de cada 4000 nacimientos y uno de cada 15 abortos espontáneos. Se podría tener en cuenta además que el 99% de los embarazos con feto 45X terminan en aborto espontáneo produciéndose, normalmente, en el primer trimestre.
Existen factores de riesgo como:
-
Edad paterna avanzada (casos de isocromosoma X)
-
Sexo femenino
-
Madre con Turner en mosaico o con delección del cromosoma X
La incidencia del síndrome no aumenta con relación a la edad materna durante la gestación. Dos tercios de las pacientes con el cromosoma X presente provienen de la madre y en un tercio de ellas proviene del padre.
No es un síndrome hereditario, no se puede evitar, ni se debe a algo que haya ocurrido durante el embarazo. No existe ningún método preventivo y las posibilidades de repetición en el siguiente feto son idénticas para una familia que ha tenido una niña con este síndrome como para otra que no la tiene, a no ser que se produzca una alteración en sus cromosomas.
Como hemos mencionado anteriormente, el síndrome de Turner es poco frecuente en varones. El género masculino deberá presentar el siguiente cariotipo:
1.- Combinación de células 45X
2.- Combinación de células 46,XY
Aunque las células 46,XY son características de varón también presentan células típicas del síndrome (45,X). Por otro lado mencionar la equivalencia del síndrome de Turner en niños 45,Y0 es nula debido a la incompatibilidad con la vida.
3.- ETIOLOGÍA DEL SÍNDROME
Existen dos teorías que explican la anomalía cromosómica:
-
Teoría meiótica: durante la formación del óvulo o los espermatozoides (gametogénesis) alguno de ellos pudo haber sufrido un error y llevar un cromosoma sexual menos. Si el óvulo o el espermatozoide ha sufrido esta pérdida cromosómica, el individuo que se forme a partir de la fertilización portará este error cromosómico.
-
Teoría mitótica: la pérdida de uno de los cromosomas no se produce en los gametos (óvulo o espermatozoide), se origina más tarde, durante el primer periodo del desarrollo embrionario (en las primeras semanas de gestación). Las investigaciones más recientes apoyan esta última teoría.
DIAGNÓSTICO:
El diagnóstico se confirma con un estudio cromosómico, un cariotipo, y además las características del fenotipo, desde el nacimiento hasta la edad adulta. El cariotipo se hace mediante una pequeña muestra de sangre venosa periférica y se tarda alrededor de cuatro o cinco días en conocer los resultados obtenidos. Los cromosomas son analizados en número suficiente para poder descartar el mosaicismo cromosómico.
El estudio con sondas del cromosoma Y se hace a las niñas con síndrome de Turner con algún signo de masculinización.
El síndrome de Turner tiene un gran parecido al síndrome de Noonan, son casi indistinguibles, pero en el caso de Noonan el cariotipo de las niñas es normal, 46XX.
El diagnóstico del síndrome lo realizan los pediatras entre la edad neonatal y la edad puberal. Un 15% de los pacientes es diagnosticado a la edad neonatal, hacia los 4 años el 8% de la población, y el resto son diagnosticadas tardíamente.
Hacia los 3 - 4 años el crecimiento se desacelera, cosa que antes había sido un crecimiento normal, y se hace más intenso el retraso hacia los 9 años
En el periodo neonatal (entre un 10 - 25 %), los niños presentan linfedema de manos y pies con pliegues cutáneos laxos en parte lateral y posterior del cuello (pterygium colli).
En el lactante se presenta un soplo cardíaco que diagnostican como coartación o estenosis aórtica.
En el periodo infantil el signo más significativo es la talla baja sin explicación.
Llegando a la pubertad la pista se da en el retraso de signos sexuales, como por ejemplo la ausencia de aumento de mama a los 13 años), espeso vello púbico o axilar e inadecuado desarrollo ovárico. También es importante destacar que tienen alteraciones gonadales y los ovarios atrofiados son sustituidos por unas cintillas fibrosas con proliferación de tejidos conjuntivos.
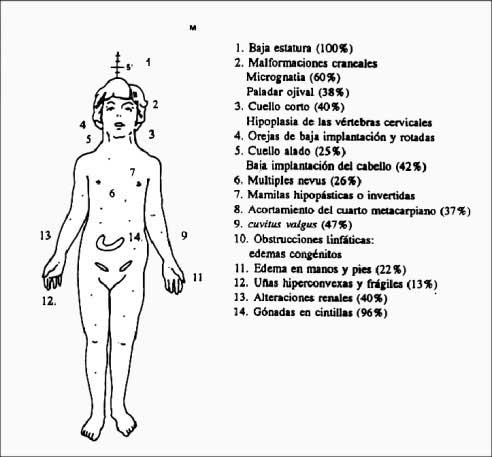
CARACTERÍSTICAS
Se las puede clasificar en:
Físicas.-
A.- De trastornos esqueléticos.-
-
Talla baja.
-
Cuello corto
-
Micrognatia (mandíbula inferior pequeña)
-
Cúbito valgo
-
4º Metacarpiano y metatarsianos cortos
-
Deformidades vertebrales con escoliosis
-
Tendencia a la obesidad
-
Genu varo
-
Tórax ancho con mamilas separadas
-
Paladar ojival y rasgos faciales de tamaño disminuido, lo que origina una resonancia en la voz (no se les extirpan los adenoides por ser beneficiosos en este caso para una emisión correcta)
B.- Alteraciones linfáticas.-
-
Pliegues cutáneos, con edema o hinchazón del dorso de manos y pies, originados a causa de problemas en la circulación
-
Cuello alado (pliegues cutáneos laxos)
-
Implantación baja del cabello (en la nuca)
-
Rotación posterior de las orejas y distorsión anatómica de la trompa de Eustaquio, provocando otitis media de repetición. A veces existe pérdida de audición.
-
Linfedema
-
Displasia de uñas
-
Alteraciones de los dermatoglifos
-
Nevus cutáneos abundantes (carnosidades en la piel)
C.- Displasias vasculares.-
-
Anomalías cardíacas
-
Anomalías renales
-
Anomalías menstruales (amenorrea habitual)
D.- Otras variantes.-
-
Estrabismo, refracción, nistagmus (bizqueo),
-
Ptosis palpebral (tendencia de uno o ambos párpados a caer)
Psicológicas.-
-
Desarrollo intelectual normal
-
C.I. manipulativo es inferior al verbal.
-
Dificultad para entender el concepto numérico y espacial
-
Dificultad en el procesamiento perceptivo-espacial:
- Escasa coordinación visomotora
-
Retraso en la madurez emocional (debido a la sobreprotección que reciben de sus padres)
-
Problemas de comportamiento social
-
Problemas de atención
-
Hiperactividad/nerviosismo
-
Baja autoestima
Fotografías
Descargar
| Enviado por: | Vereña |
| Idioma: | castellano |
| País: | España |
Todos los derechos reservados.