Salud
Serotonina
UNIVERSIDAD GUADALAJARA LAMAR.
“SEROTONINA”
GUADALAJARA, JAL., A 26 DE NOVIEMBRE DE 2004.
INTRODUCCIÓN:
La indolalquilamina 5-hidroxitriptamina (5-HT; serotonina) fue inicialmente identificada por el interés de sus efectos cardiovasculares.
Desde mediados del siglo XIX se sabe que actúa sobre la musculatura lisa de los vasos sanguíneos y por tanto posee un importante efecto hipertensor.
A principios del siglo XX, las plaquetas fueron identificadas como la fuente de esta sustancia. A finales de la década de los 40, Page y sus colaboradores, aislaron y caracterizaron esta sustancia tónica del suero (serum; de aquí se origina el nombre de serotonina).
La combinación del grupo hidroxilo en la posición 5 del núcleo indol y una amina nitrogenada primaria actuando como aceptador de un protón del pH fisiológico, hace de la 5-HT una sustancia hidrofílica. Como tal, no traspasa la barrera hematoencefálica fácilmente. Así, su descubrimiento en el cerebro en 1953 por Twarog y Page indicó que la 5-HT estaba siendo sintetizada en el cerebro.
La observación casi simultanea de que la droga psicodélica Dietilamida del Ácido Lisérgico (LSD) antagoniza una respuesta producida por 5-HT (aún cuando la respuesta fuera la contracción del músculo liso gastrointestinal) confirmó la idea de que la 5-HT era un producto de nuestro cerebro y tiene importantes efectos conductuales.
BIOQUIMICA
| Serotonina | |
| Nombre Químico | 5-Hidroxi-triptamina o |
| Fórmula Química | C10H12N2O |
| Masa Molecular | 176.22 g/mol |
| Número de CAS | 50-67-9 |
| Símiles | NCCC1=CNC2=C1C=C(O)C=C2 |
{kind=link}
SEROTONINA
La indolalquilamina 5-hidroxitriptamina (5-HT; serotonina) resulta de la combinación del grupo hidroxilo en la posición 5 del núcleo indol y una amina nitrogenada primaria actuando como aceptador de un protón del pH fisiológico, esto hace de la 5-HT una sustancia hidrofílica. Como tal, no traspasa la barrera hematoencefálica fácilmente.
No todas las células que contienen 5-HT lo sintetizan. Las plaquetas no sintetizan 5-HT; acumulan la 5-HT del plasma por un mecanismo de transporte activo que se encuentra en la membrana de plaquetas. El paso inicial en la síntesis de serotonina es el transporte facilitado del aminoácido L-triptófano de la sangre hasta el cerebro. Otros aminoácidos neutros (fenilalanina, leucina, metionina) son transportados dentro del cerebro por el mismo mensajero.
Las neuronas serotoninérgicas contienen la enzima triptófano-hidroxilasa, que convierte el triptófano en 5-hidroxitriptófano (5-HTP); su distribución en el cerebro es similar a la de la propia 5-HT. La enzima requiere tanto de oxigenación molecular del cofactor biopteridina
La otra enzima implicada en la síntesis de serotonina es el descarboxilasa de los aminoácidos L-aromático (aminoácido descarboxilasa: AADC), que convierte 5-HTP en 5-HT. Esta enzima está presente no sólo en las neuronas serotoninérgicas sino también en las neuronas catecolaminérgicas, donde convierte 3,4-dihidroxifenilalanina (DOPA) a dopamina.
La hidroxilación inicial del triptófano parece ser el peldaño limitante en la síntesis de serotonina más que la descarboxilación de 5-HTP. La evidencia en apoyo para este punto de vista incluye el hecho de que 5-HTP se encuentra sólo en pequeñas cantidades en el cerebro, presumiblemente porque es descarboxilado casi tan rápidamente como se forma.
{kind=link}
En 1964, Dahlstrom y Fuxe, usando la técnica de histofluorescencia de Falck-Hillarp, observó que la mayoría de cuerpos serotoninérgicos fueron encontrados en grupos de los cuerpos celulares previamente designados por Taber, Brodal, y Walberg como el núcleo de Raphé. Dahlstrom y Fuxe describieron nueve grupos de cuerpos celulares que contienen serotonina, a los que ellos designaron desde B1 hasta B9, y que se corresponden en su mayor parte con el núcleo de Raphé.
El grupo más grande de células serotoninérgicas es el grupo B7 contiguo a un grupo más pequeño de células serotoninérgicas, B6. Los grupos B6 y B7 son a menudo considerados conjuntamente como el núcleo dorsal de Raphé, con B6 siendo su extensión caudal. Otro grupo de cuerpos celulares serotoninérgicos prominente, es el B8 que corresponde al núcleo medio de Raphé, también llamado el núcleo central superior. El grupo B9, parte del tegmento ventrolateral del puente y del cerebro medio. Las proyecciones serotoninérgicas ascendentes, que inervan el córtex y otras regiones del cerebro anterior vienen desde el Raphé dorsal, Raphé medio, y el grupo celular B9. El otro núcleo de Raphé B1 a B5 está situado más caudalmente y contiene un número bajo de células serotoninérgicas.
Dos caminos principales serotoninérgicos de ascenso emergen del núcleo de Raphé del cerebro medio al cerebro anterior: el camino dorsal periventricular y las radiaciones ventral tegmental. Ambos convergen en el hipotálamo caudal donde se unen al haz medial del cerebro anterior (MFB).
Los núcleos de Raphé dorsal y medial dan salida a múltiples paquetes distintos de axones que forman caminos separados para diferentes regiones cerebrales. Estructuras funcionalmente relacionadas en el cerebro son inervadas por el mismo grupo de neuronas serotoninérgicas. Por ejemplo, el hipocampo y el séptum (estructuras límbicas) parecen estar inervadas predominantemente por neuronas del Raphé medial, mientras el estriado y la sustancia nigra (sistema de los ganglios basales que median la actividad motora) son inervados por el Raphé dorsal. Los dos núcleos de Raphé mandan proyecciones neuronales solapadas al neocórtex. Además, células dentro del Raphé dorsal y medial son organizadas en zonas particulares o grupos que mandan axones a áreas específicas del cerebro como el córtex o hipocampo. Por ejemplo, el córtex frontal recibe fuerte inervación de subregiones rostrales y laterales del núcleo de Raphé dorsal. Estructuras relacionadas funcionalmente en el cerebro pueden también ser inervadas por las mismas neuronas individuales. Las neuronas serotoninérgicas mandan axones colaterales a más de una región cerebral, a menudo a las áreas terminales que están funcionalmente relacionadas como el córtex entorrinal y el hipocampo.
La síntesis de 5-HT puede aumentar de forma marcada, bajo condiciones que requieren un continuo suministro del neurotransmisor. La plasticidad es un concepto importante en neurobiología. En general, se refiere a la capacidad de los sistemas neuronales para ajustarse a demandas a corto o largo plazo sobre su actividad o funcionamiento. Muchos procesos contribuyen a la plasticidad neuronal.
Uno, es la capacidad para aumentar la proporción de síntesis del neurotransmisor y liberación en respuesta a un incremento de la actividad neuronal. El aumento de la síntesis resulta desde la conversión realizada del triptófano en 5-HTP y tiene una absoluta dependencia del Ca2+ extracelular.
Por contraste, las situaciones que requieren aumentos en la síntesis y emisión de 5-HT a largo plazo, resultan en la síntesis de la proteína triptófano-hidroxilasa. Por ejemplo, la parcial pero sustancial destrucción de las neuronas centrales serotoninérgicas resulta en un aumento de la síntesis de 5-HT en terminales residuales. El aumento de la síntesis de 5-HT resulta de más triptófano-hidroxilasa que está presente en los terminales residuales.
Como con otros transmisores amino biogénicos, la 5-HT es almacenada primariamente en vesículas y es liberada por un mecanismo exocitótico.
En algunos aspectos éstas vesículas que almacenan 5-HT se parecen a aquellas que almacenan catecolaminas (CAs). Por ejemplo, los medicamentos como reserpina y tetrabenzina, que inhiben la actividad del transportador localizado a la membrana vesicular, reducen el contenido celular de 5-HT así como CAs. Esta reducción inducida por los medicamentos, del contenido de 5-HT, muestra que el almacén vesicular de 5-HT es necesario para proteger la indolalquilamina de degradación intraneuronal por la monoaminooxidasa.
En otros aspectos, las vesículas que almacenan 5-HT son diferentes de aquellas que almacenan CAs. En contraste con las vesículas que contienen CAs, no hay prácticamente ATP en las vesículas serotoninérgicas. También, las vesículas sinápticas serotoninérgicas, pero no los gránulos de cromafín, contienen una proteína específica que se adhiere a la 5-HT con gran afinidad. Esta proteína adherente a la serotonina (SPB) desaparece del cerebro anterior tras una lesión del núcleo de Raphé, indicando que la SPB está contenida en neuronas serotoninérgicas. La SPB es liberada sólo con serotonina por un proceso dependiente del Ca2+.
Como se esperaba, los terminales serotoninérgicos hacen los contactos usuales sinápticos especializados con las neuronas objetivo y liberan serotonina siguiendo la estimulación nerviosa. En la mayoría de las áreas del sistema nervioso central de los mamíferos, hay al menos algunos lugares donde la 5-HT es liberada y no se ha encontrado evidencia sobre la especialización sináptica. En este caso, el neurotransmisor es liberado y difundido a cierta distancia. El porcentaje de terminales 5-HT asociados con especializaciones sinápticas, varía en regiones cerebrales particulares. La apariencia de contactos sinápticos especializados sugiere asociaciones relativamente estables y fuertes entre una neurona presináptica y su objetivo. A la inversa, la ausencia de especialización sináptica implica una interacción dinámica y quizás menos específica, con las neuronas objetivo. En este caso, la 5-HT puede actuar como un neuromodulador. La actividad de 5-HT en la sinapsis se termina, primariamente, por su recogida en terminales serotoninérgicos.
Los efectos sinápticos de muchos aminoácidos y neurotransmisores monoaminérgicos, incluida la 5-HT, son terminados por unión de estas moléculas a proteínas específicas de transporte. El sistema de transporte para la 5-HT esta localizado en las neuronas serotoninérgicas. Las células gliales también parecen ser capaces de coger 5-HT por un sistema de transporte de gran afinidad.
La estructura del transporte de serotonina es bastante diferente de la estructura de los receptores asociados a la proteína. Los medicamentos que son inhibidores selectivos de la recogida de 5-HT, como la fluoxetina o sertralina, son ampliamente usados como antidepresivos. La clomipramina, que tiene una selectividad moderada in vivo para inhibir la recogida de 5-HT frente a aquellos de NA, es usada para el tratamiento del trastorno de ansiedad, trastorno obsesivo-compulsivo. Producen inhibición competitiva de la recogida de 5-HT, y una simple proteína parece ser responsable tanto de la unión de estos medicamentos como de la recogida de 5-HT.
El camino primario catabólico para la 5-HT es la desaminación oxidativa por la enzima monoaminooxidasa.
La monoaminooxidasa (MAO) convierte la serotonina en 5-hidroxi-indoleacetaldehído, y este producto es oxidado por una aldehído deshidrogenasa dependiente de NAD+ para formar ácido 5-hidroxi-indolacético (5-HIAA).
Hay al menos dos isoenzimas de MAO, denominadas como tipo A y tipo B. Estas isoenzimas son completamente flavoproteínas de membranas mitocondriales externas en neuronas, glía y otras células. Existen inhibidores selectivos de cada forma de MAO, ej., clorgilina o maclobemida para el tipo A o deprenil para el tipo B.
El cerebro humano contiene más tipo B, que tipo A. Es interesante que los cuerpos celulares de la serotonina contienen predominantemente MAO tipo B, así los nervios serotoninérgicos (al menos, los somas) contienen la forma de MAO (tipo B) que no metaboliza preferentemente 5-HT. Esto ha llevado a la hipótesis de que la MAO tipo B en las neuronas serotoninérgicas, impide a la célula la acumulación de varios substratos naturales (ej., dopamina) que puede interferir con el almacenamiento, liberación y recogida de 5-HT.
RECEPTORES PARA LA SEROTONINA:
Estudios farmacológicos y fisiológicos han contribuido a la definición de muchos subtipos de receptores para serotonina. Inicialmente se diferenciaron dos receptores diferentes de 5-HT en el íleon, llamados receptores D (bloqueado por dibencilina) y M (bloqueado por morfina). El receptor D se pensó que estaba en el músculo liso del íleon mientras que el receptor M, se consideró que estaba en la estructura ganglionar.
El desarrollo del ensayo de unión al radioligando fue propuesto por Pertoutka y Snyder en 1979 para etiquetar dos clases de receptores serotoninérgicos en el cerebro. Los lugares de unión con alta afinidad por [3H]5-HT fueron designados como receptor 5-HT1; los lugares de unión etiquetados con alta afinidad por [3H]espiperona fueron denominados como receptor 5-HT2.
La unión de [3H]5-HT a los receptores 5-HT1 fue desplazada por la espiperona de forma bifásica, sugiriendo que el llamado receptor 5-HT1 podía ser una población heterogénea de receptores. El lugar de unión [3H]5-HT que muestra alta afinidad para espiperona fue llamado subtipo 5-HT1A, mientras el componente de unión [3H]5-HT que mostraba baja afinidad para espiperona fue llamado el subtipo 5-HT1B. Se encontró una alta densidad de lugares de unión para [3H]5-HT en el plexo coroideo. Estos sitios de unión a [3H]5-HT fueron denominados subtipo 5-HT1C. Un cuarto lugar de unión para [3H]5-HT fue identificado en el cerebro bovino y fue llamado receptor 5-HT1D. El receptor 5-HT1D fue identificado en cerebros de especies desprovistas de receptor 5-HT1B.
El receptor M de Gaddum y Picarrelli, originalmente descrito en el íleon de cobayas. Bradley y asociados han renombrado a este receptor como 5-HT3. Un subtipo adicional de receptor serotoninérgico ha sido descrito, el receptor 5-HT4.
Tanto en el hipocampo como en el núcleo de Raphé, los receptores 5-HT1A están asociados a la apertura de los canales K+, presumiblemente de forma directa a través de una proteína G. En las áreas del campo terminal como el hipocampo, los receptores 5-HT1A están también asociados mediante una proteína G a la inhibición de la actividad de la adenilciclasa. El receptor 5-HT1A es clasificado como estando asociado a ambos la estimulación y la inhibición de la adenilciclasa, siempre en la misma región cerebral.
En la sustancia nigra, demostrada por estudios de unión de radioligandos, una alta densidad de receptores 5-HT1B y 5-HT1D, estos receptores serotoninérgicos están asociados a la inhibición de adenilciclasa a través de la proteína G.
Los receptores 5-HT1C y 5-HT2 están asociados a través de la proteína G a la estimulación de hidrólisis de fosfoinositol (PI)
El receptor 5-HT3 es un ion ligado a la apertura de canal, es un canal iónico tal que la reacción provocada por su activación no es mediada por segundo mensajero o a través de proteínas G.
El receptor 5-HT4 en neuronas del colículo e hipocampo está asociado a la estimulación de la actividad de la adenilciclasa y a la inhibición de canales K+. Se ha demostrado que la inhibición de canales K+ en neuronas del colículo implica la producción de AMPc y la activación de proteínoquinasa A dependiente de AMPc. A pesar de que el sistema del segundo mensajero asociado con el receptor 5-HT4 es AMPc, esto parece ser si otro mecanismo de transducción también se asocia a los receptores 5-HT4.
Los muchos subtipos de receptores para serotonina no son sólo distinguibles por su farmacología y sistemas de segundo mensajero, sino también por su localización en el SNC.
IMPLICACIONES FUNCIONALES DE LA SEROTONINA
Los receptores 5-HT1A están presenten en alta densidad en el hipocampo, séptum, amígdala, hipotálamo y neocórtex. La destrucción de neuronas serotoninérgicas con la neurotoxina 5,7-dihidroxitriptamina (5,7-DHT), no reduce el número de receptores 5-HT1A en áreas del cerebro anterior, lo que indica que los receptores de 5-HT1A están localizados postsinápticamente en éstas regiones del cerebro. Muchas de estas áreas terminales del campo serotoninérgico, son componentes del sistema límbico, el curso que se piensa que está implicado en la modulación de la emoción. La presencia de receptores 5-HT1A en alta densidad en el sistema límbico, indica que los efectos propuestos de 5-HT o medicamentos serotoninérgicas en los estados emocionales, pueden estar mediados por los receptores 5-HT1A.
El receptor 5-HT1B en ratas y ratones y el receptor 5-HT1D en cerebro bovino y humano está situado en alta densidad en los ganglios basales, particularmente en globo pálido, y la sustancia nigra. Los receptores 5-HT1B y 5-HT1D están situados postsinápticamente donde pueden modular la liberación de otros neurotransmisores, como acetilcolina. La presencia de estos receptores en alta densidad en los ganglios basales, aumenta la interesante posibilidad de que estos receptores puedan estar implicados en trastornos del cerebro que implican a los ganglios basales, como la enfermedad de Parkinson.
Los receptores 5-HT1C están presentes en alta densidad en el plexo coroideo. Se ha propuesto que la activación inducida por 5-HT de los receptores 5-HT1C podría regular la composición y volumen del líquido cefalorraquídeo.
Una alta densidad de los receptores 5-HT2 se encuentra en muchas áreas del córtex. En el neocórtex, estos receptores están concentrados en las capas I y V. Los receptores 5-HT2 también se encuentran en una particular alta densidad en el claustrum, una región que está conectada al córtex visual, a partes del sistema límbico, y a los ganglios basales y al núcleo olfatorio.
Los receptores 5-HT3 inicialmente parecen estar confinados a neuronas periféricas, donde median acciones despolarizantes de 5-HT y modulan la liberación del neurotransmisor. Los receptores 5-HT3 se encuentran en alta densidad en ganglios y nervios periféricos (ganglio superior cervical y nervio vago) así como en la sustancia gelatinosa de la médula espinal. Su situación en el cordón espinal y médula sugiere que 5-HT puede modular mecanismos nocioceptivos por medio del receptor 5-HT3. La mayor densidad del receptor 5-HT3 en el cerebro, es en el área postrema, el lugar de la zona de disparo del receptor químico.
El receptor 5-HT4, originalmente caracterizado por la medida en la producción AMPc de neuronas coliculares cultivadas de ratones, ha sido también localizado en el hipocampo.
Muchos de los subtipos de receptores de serotonina no parecen experimentar cambios reguladores compensatorios, como en un principio describieron Cannon y Rosenblueth en 1949 para receptores colinérgicos nicotínicos en el periférico.
Clásicamente, una disminución en la exposición de un tejido a su transmisor endógeno, lleva a una reacción hipersensible o exagerada a agonistas exógenos, lo cuál puede justificarse por un incremento en la densidad de receptores postsinápticos para el transmisor (regulación al alza). A la inversa, la exposición aumentada de un tejido a agonistas, puede, con el tiempo, resultar en una reacción disminuida al agonista (desensibilización), que puede ser debida a un decremento en la densidad del receptor (regulación a la baja).
La administración crónica o repetida de medicamentos antidepresivos (ej., inhibidores de la MAO o inhibidores de la recogida de serotonina) o receptores agonistas de 5-HT1A en ratas de laboratorio, produce una desensibilización de reacciones conductuales y electrofisiológicas que se creía eran mediadas por receptores 5-HT1A. Lesionando neuronas serotoninérgicas se produce un incremento de respuestas conductuales y electrofisiológicas. Sin embargo, estos tratamientos, no producen cambios en los receptores 5-HT1A medidos con ensayos de unión. Algunos investigadores han propuesto la disminución de la inhibición de adenilciclasa mediada por el receptor 5-HT1A.
Las lesiones de neuronas serotoninérgicas no causa cambios detectables en receptores 5-HT1B en áreas del cerebro anterior y se ha propuesto que causan regulación a la alta o a la baja o no afectan a la densidad de los receptores 5-HT1B en la sustancia negra.
Siguiendo la lesión de neuronas serotoninérgicas con neurotóxico, el receptor 5-HT1C mediado por hidrólisis PI en el plexo coroideo; aumenta, y estos receptores experimentan supersensibilidad a la denervación.
Los receptores 5-HT2, tampoco producen cambios en la exposición al agonista en la forma clásica. Específicamente, no se observa cambio en la densidad del receptor 5-HT2 tras la lesión de neuronas serotoninérgicas o tras la deplección de almacenes serotoninérgicos. Así, parece que ni el receptor 5-HT2 ni el camino de su segundo mensajero son regulados por un decremento en la exposición al neurotransmisor. Tras la administración de agonistas alucinógenos del receptor 5-HT2, la administración crónica de inhibidores selectivos de recogida de serotonina, o antagonistas del receptor 5-HT2, la hidrólisis PI mediada por el receptor 5-HT2 se convierte en desensibilizada y los receptores 5-HT2 regulan a la baja.
Los receptores 5-HT3, localizados en neuronas en el sistema nervioso central y periférico, median rápido, reacciones excitatorias (depolarización de la membrana) a la serotonina. Como muchos otros receptores que están directamente asociados a un canal iónico, el receptor 5-HT3 exhibe una rápida desensibilización tras la exposición sostenida al agonista.
La serotonina está entre los muchos neurotransmisores que participan en el control hipotalámico de la secreción pituitaria, particularmente en la regulación de prolactina, adrenocorticotropina (ACTH), y hormona del crecimiento. La medida de estas reacciones endocrinas tras la administración de medicamentos que incrementan la función de la serotonina en el cerebro, proporciona uno de los pocos métodos actualmente disponible para evaluar dicha función en humanos. Por ejemplo, la administración del precursor de serotonina, L-triptófano, aumenta las concentraciones en plasma de prolactina y hormona de crecimiento. Cuando se administra a humanos agonistas serotoninérgicos que estimulan los receptores 5-HT1A, 5-HT1C y 5-HT2, también aumentan en plasma las concentraciones de ACTH, prolactina y hormona de crecimiento. La reacción neuroendocrina en humanos, al agonista no selectivo del receptor serotoninérgico m-CPP (m-clorofenilpiperazina), o a L-triptófano, ha sido utilizada clínicamente para evaluar el funcionamiento del sistema central serotoninérgico en pacientes con trastornos psiquiátricos.
La investigación en gatos, ha implicado a la serotonina en el sueño y en estados de activación (arousal). Las neuronas serotoninérgicas en el núcleo de Raphé dorsal muestran un cambio dramático en la actividad a lo largo del ciclo de sueño-vigilia-activación. Bajo condiciones de vigilia tranquila, las neuronas serotoninérgicas exhiben una actividad lenta, imitando a un reloj, la cuál muestra una disminución gradual conforme el animal va volviéndose somnoliento y entra en el sueño de ondas lentas. Un decremento en la regularidad del disparo acompaña esto, sobre todo disminución de la actividad neuronal. Durante el sueño REM, (movimientos rápidos de ojos), la actividad de estas neuronas cesa. En respuesta al estimulo activado, la tasa de disparo de estas neuronas serotoninérgicas aumenta. Un estímulo auditivo (golpe) o visual (destello), produce una excitación de las neuronas serotoninérgicas del Raphé dorsal, seguida por una inhibición. Sin embargo, exponiendo un gato a estresores ambientales como un sonido fuerte o la visión de un perro, aunque produce una activación simpática fuerte y una reacción conductual típica, no altera la tasa de disparo de estas neuronas serotoninérgicas. Ya que la actividad tónica de neuronas serotoninérgicas parece variar de forma general en asociación con un estado conductual y no asociado con ninguna reacción conductual específica, Jacobs y colaboradores han propuesto que el papel de las neuronas centrales serotoninérgicas es coordinar la actividad del sistema nervioso, fijar el tono de actividad en conjunción con el nivel de activación del organismo.
La serotonina también parece estar implicada en la regulación de ritmos circadianos. El núcleo supraquiasmático del hipotálamo genera ciclos electrofisiológicos y metabólicos que repite aproximadamente cada 24 horas. Generalmente, este ritmo esta sincronizado al foto período del ambiente, también de alrededor de 24 horas. Ha sido postulada una contribución serotoninérgica a la regulación del ritmo circadiano porque el quiasmático recibe una inervación serotoninérgica muy densa del núcleo de Raphé desde el cerebro medio. Muy poco se conoce, no obstante, sobre la función de esta densa entrada serotoninérgica. Las lesiones de neuronas serotoninérgicas en animales de laboratorio han sido presentadas por algunos investigadores, pero no por todos, para romper los ritmos locomotores o que resultan en la pérdida del ritmo diario de corticosterona. Cuando se aísla in vitro, el quiasmático continua produciendo ritmos en el metabolismo de 24 horas, secretando vasopresina y realizando actividad eléctrica espontánea, indicando que las funciones conservadas de tiempo circadiano o actividad de establecedor del ritmo son características endógenas del quiasmático. El agonista no selectivo de 5-HT, quipazina se ha mostrado que reajusta o traslada el ritmo de la actividad eléctrica espontánea de células simples registradas extracelularmente en el quiasmático aislado en partes de cerebro. Estos resultados sugieren que el establecedor de ritmo circadiano del quiasmático o reloj es modulado por estimulación de los receptores serotoninérgicos en el mismo y que las proyecciones serotoninérgicas al quiasmático pueden modular la fase del quiasmático en animales intactos.
La investigación neuroquímica, se ha focalizado en cómo afecta la alimentación a las concentraciones de triptófano en el cerebro y en la síntesis y disponibilidad de serotonina, mientras que la investigación farmacológica ha estado basada en el control del apetito por medio de medicamentos serotoninérgicos. La administración de agonistas serotoninérgicos no selectivos indirectos, como la fenfluramina, cuyo trabajo es liberar serotonina, o 5-hidroxitriptófano, un precursor de síntesis de serotonina, en ratas de laboratorio, disminuye el apetito. De estos datos, se ha inferido que la serotonina inhibe la toma de comida. Los agonistas serotoninérgicos activando los receptores postsinápticos 5-HT1C y 5-HT1B también disminuyen el apetito. Los inhibidores selectivos de recogida de serotonina tienen efectos anoréxicos, también presumiblemente por acciones fisiológicas de intensificación de serotonina endógena. Por contraste, las dosis pequeñas de agonistas selectivos de 5-HT1A aumenta la toma de comida en ratas. El aumento de consumición de comida, puede ser debido a actividad agonista de autorreceptores serotoninérgicos en el núcleo de Raphé. La activación de receptores somatodendríticos 5-HT1A podría esperarse que inhiba el disparo neuronal serotoninérgico y la liberación de serotonina. Los efectos hipofágicos de la fenfluramina o agonistas de 5-HT1 son más pronunciados en ratas hembras, un efecto de potencial relevancia para los trastornos de la alimentación en humanos, como anorexia nerviosa y bulimia nerviosa, que tienen una mayor tasa de incidencia en mujeres jóvenes que en hombres jóvenes.
El papel de la serotonina (5-HT) en el SNC está completamente ligado al de la NA, ya que interviene en la regulación de la vigilancia, en el proceso activo del sueño, la atención, en los procesos motivacionales y en la regulación de los estados de ánimo. Por otra parte, no debemos olvidar que el control de entrada del dolor parece depender de la liberación de serotonina, que facilita la producción de endomorfinas medulares.
Todo este enorme papel se realiza fundamentalmente a través de una localización sucesiva de estructuras, los núcleos del Raphé. Las proyecciones de estos núcleos a través del fascículo medio del telencéfalo suelen ser inhibitorias, de ahí que el papel regulador de la actividad de las catecolaminas sea inseparable del de la serotonina, hasta tal punto que para describir las áreas catecolaminérgicas del tronco encefálico se utiliza la terminología A y, por ejemplo, el locus ceruleus es la estructura A-6. Pues bien, para los distintos núcleos serotoninérgicos se emplea la terminología B, y así llamamos B1 al núcleo del Raphé pálido, B2 al Raphé oscuro y B3 al Raphé magnus, como núcleos bulbares que se caracterizan por las proyecciones descendentes y, por tanto, medulares coincidiendo con las estructuras A1 y A2 noradrenérgicas. Los núcleos B4, B5 y B6 son los núcleos pontinos del Raphé. Los grandes núcleos B7, B8 y B9, que son los núcleos dorsal y medianos del Raphé, son las estructuras más rostrales, y desde ellos se ofrecen las proyecciones serotoninérgicas ascendentes hacia el hipotálamo, el tálamo, los núcleos grises basales, el sistema límbico y la corteza frontal.
La destrucción de los núcleos del Raphé, o la administración de una sustancia como la dihidroxitriptamina, conduce a un incremento de la actividad nerviosa. Sin embargo, también hay una serie de situaciones en las que la serotonina en lugar de inhibir, excita, demostrándose esta acción con los efectos activadores autónomos y motores, y no olvidemos los efectos alucinógenos de los propios agonistas de la serotonina que, mediante una actuación sobre los receptores presinápticos, ofrecen una consecuencia de hiperactividad típica de las alucinaciones táctiles y visuales.
REGULACIÓN DEL SUEÑO
En la actualidad, la idea que prevalece es que no existe un único centro del SNC que controle el sueño, sino que existe un número de sistemas o centros interconectados, que se activan mutuamente o se inhiben unos a otros, a través de los neurotransmisores y neuromoduladores, los principales son:
Serotonina
Muchos estudios apoyan la participación de la serotonina (5-HT) en el sueño, ya que la administración del L-triptófano induce al sueño y se le llama hipnótico natural. La síntesis y liberación de serotonina dependen de la disponibilidad de aminoácidos precursores del L-triptófano (de 1 a 15g), que reduce la latencia de sueño y los despertares nocturnos. Por el contrario, la deficiencia de L-triptófano se asocia a una reducción del sueño MOR. Sin embargo no se utiliza a nivel clínico, porque se le relacionó con el síndrome de mialgia eosinofílica.
Lesiones en el núcleo dorsal del rafe se acompañan de agotamiento de la serotonina e insomnio que dura días. La administración de p-clorofenilalanina inhibe a la hidrosilasa de triptófano y también produce insomnio en el gato durante 3 o 4 días. Este insomnio puede revertirse con la administración de 5-hidroxitriptófano, otro precursor de la serotonina que evita el sitio de inhibición enzimática de la p-clorofenilalanina. Por otro lado, la serotonina regula la aparición de espigas ponto-geniculo-occipitales (PGO), las cuales aparecen segundos antes del inicio del sueño NMOR y durante todo el tiempo que dura éste.
Los niveles de serotonina varían a lo largo del día, así como el número y afinidad de sus receptores. Los 5-HT tienen niveles máximos durante el día y disminuye durante la noche; los receptores muestran una curva inversa, ya que son más abundantes y afines durante la noche. En experimentos de deprivación de sueño se detectó aumento de los niveles de serotonina, pero los receptores no se modificaban.
Noradrenalina
En un estado de activación intervienen dos áreas cerebrales, cada una con un neurotransmisor propio, son el locus coeruleus y la sustancia negra. Las neuronas que contienen noradrenalina, cuyos cuerpos celulares se localizan en el locus coeruleus, están muy activas durante la vigilia, pero se encuentran silentes durante el sueño MOR. Las lesiones en esta área producen hipersomnia (exceso de sueño), aumentando tanto el sueño de ondas lentas como el sueño MOR (Jones, Bobillier y Jouvet, 1969). En seres humanos, la estimulación eléctrica del locus coeruleus, altera profundamente todos los parámetros del sueño.
Los estudios farmacológicos ofrecen resultados contradictorios. La inhibición de la hidrosilasa de tirosina con alfa-metil-paratoroxina (AMPT), produce un descenso de la noradrenalina y la dopamina con hipersomnia y disminución de la latencia hasta el NMOR. Estudios posteriores que analizaron el papel de los receptores alfa1, alfa2 y adrenérgicos-b, mostraron que hay una interacción importante entre ellos en el mantenimiento y equilibrio del ciclo sueño-vigilia. La administración periférica de clonidina, un agonista alfa2 que disminuye la liberación de noradrenalina, disminuye el sueño NMOR y MOR; la aplicación local de clonidina en el locus coeruleus suprime el NMOR. El empleo de un antagonista alfa1 como la fenoxibenzamina también reduce el NMOR. También es probable que la noradrenalina participe en la aparición del ritmo theta, ya que la administración de clonidina y p-aminoclonidina induce la actividad theta hipocámpica. En general, los agentes que aumentan la disponibilidad de la noradrenalina en la hendidura sináptica suprimen el NMOR, no obstante, hay excepciones como la clonidina, la cual disminuye la liberación de este neurotransmisor y también suprime el MOR.
Dopamina
El segundo sistema de activación se localiza en la sustancia negra, cuyas neuronas utilizan un neurotransmisor catecolaminérgico, la dopamina. Este centro está implicado en la coordinación motora y en el tono muscular postural. Los efectos de las anfetaminas y la cocaína sugieren el papel este neurotransmisor en el mantenimiento de la vigilia. Las anfetaminas son estimulantes que aumentan la vigilia y disminuyen el NMOR. Los efectos de las anfetaminas pueden bloquearse con pimocida, un antagonista que se utiliza como neuroléptico en la práctica clínica.
Los niveles de dopamina son altos durante la vigilia y el recambio disminuye en la transición del estado de despierto a dormido. En estudios animales, la apomorfina (un antagonista dopaminérgico), origina un predomino de la vigilia a expensas del sueño, sobre todo del NMOR. Se obtiene el efecto contrario con la administración de pimocida y otros neurolépticos (sulpiride y haloperidol) antagonistas de los receptores dopaminérgicos D2.
En general, las sustancias que incrementan la dopamina cerebral producen activación y vigilia, por el contrario los bloqueadores de la dopamina como la pimocida y las fenotiacidas, tienden a incrementar el tiempo de sueño.
Acetilcolina
La acetilcolina cerebral también está implicada en la regulación del sueño, en particular en la producción del MOR. En humanos, los agentes agonistas colinérgicos como la fisostigmina, arecolina, RS-86 y pilocarpina inducen el NMOR; por el contrario la escopolamina, un antagonista no selectivo y el biperidén selectivo para los receptores muscarínicos M1, tienen efectos opuestos.
Parece que un grupo de neuronas colinérgicas en la formación reticular de la protuberancia se encargan del inicio y mantenimiento del NMOR. La administración sistemática de atropina produce ondas lentas de gran amplitud, mientras que la infusión local de agonistas en la formación reticular aumenta la desincronización cortical y la intensidad del despertar conductual.
La actividad theta es uno de los componentes tónicos del NMOR, el sistema colinérgico es un mediador parcial de éste, de manera que la atropina puede modificar elementos de la actividad theta. La atonía también tiene componente colinérgico, ya que la inhibición de los músculos antigravitatorios proviene de la activación de grupos de neuronas no aminérgicas que se localizan en la periferia del locus coeruleus (peri-LC-alfa). La infusión de carbacol dentro del tegumento pontino induce cataplexia rápida en el gato.
También se asocia la acetilcolina (Ach) con la actividad PGO. La administración de atropina reduce significativamente los trenes de estas espigas, sin embargo también neuronas noradrenérgicas y serotoninérgicas tienen influencia inhibitoria sobre las espigas PGO. La administración de reserpina, que bloquea el almacenamiento de monoaminas e índoles, da lugar a la aparición de PGO en vigilia y en otras fases de sueño diferentes a la de MOR.
Las alteraciones en la actividad colinérgica central se asocian a cambios de sueño observados en el Trastorno Depresivo Mayor, que muestran anormalidades importantes en los patrones del MOR, entre ellas: acortamiento de la latencia MOR (60´), incremento del porcentaje de sueño MOR, y un cambio en la distribución del mismo desde la primera mitad de la noche hasta la última. La administración de un agonista muscarínico, a pacientes deprimidos durante el primer o segundo periodo NMOR provoca un rápido inicio del sueño MOR. Así, la depresión puede asociarse a una hipersensibilidad subyacente a la acetilcolina (Kaplan y cpls., 1996).
Las sustancias que reducen el sueño MOR, como los antidepresivos, producen efectos beneficiosos sobre la depresión, además casi la mitad de los pacientes con un trastorno depresivo mayor experimentan mejorías temporales cuando se les depriva del sueño. Por el contrario, la reserpina, que es una de las pocas sustancias que aumentan el sueño MOR, también produce depresión.
Los pacientes con Demencia tipo Alzheimer presentan alteraciones en el sueño caracterizadas por una reducción del MOR y sueño de ondas lentas. La pérdida de neuronas colinérgicas en el cerebro anterior se implican en estos cambios.
Adenosina
La adenosina es un nucleósido de purina tiene efectos sedantes e inhibitorios sobre la actividad neuronal. La cafeína disminuye el sueño precisamente por el bloqueo del receptor de adenosina. La adenosina aumenta el sueño NMOR (sobre todo en el estadio IV) y también el NMOR. Cuando se aplica un inhibidor de la desaminasa de adenosina (desoxicoformicina) se incrementa el NMOR. Se observó el mismo efecto con el precursor de la adenosina, el S-adenosil homocisteína.
Aún no se identifica el papel de la adenosina en la vigilia, pues los receptores de adenosina A1 tras la privación de NMOR estaban elevados, sin embargo los niveles de adenosina a las 48 horas de abstinencia no estaban altos.
Histamina
El papel de la histamina en el ciclo vigilia-sueño se identificó en base a observaciones farmacológicas en el mantenimiento de la vigilia y del efecto sedante de los antagonistas de los receptores H1, que en los humanos producen somnolencia. Estos antihistamínicos acortan la latencia del sueño, pero no modifican significativamente el sueño nocturno, por ello es un componente frecuente en los inductores al sueño. El principal problema es que crea una tolerancia rápida al efecto hipnótico. Los medicamentos que actúan sobre los receptores H2 no parecen tener efecto sobre la vigilia, pero aumentan la cantidad de sueño delta.
Se supone que hay una vía histaminérgica ascendente que proporciona inervación a la corteza, cuerpo estriado, hipocampo y amígdala, cuyos cuerpos celulares se localizan en el hipotálamo posterior y regiones difusas del mesencéfalo, que en el hombre, participarían del control de la vigilia y en el equilibrio entre vigilia y sueño.
GABA
La principal evidencia que relaciona al Ácido gammaaminobutírico (GABA) con los mecanismos del sueño son las asociaciones entre los receptores gabaérgicos y las benzodiacepinas, que hoy día son los medicamentos hipnóticos de mayor prescripción. La administración de l-cicloserina inhibe la destrucción del GABA, y tiene un efecto similar al de las benzodiacepinas en el sueño, con la diferencia que las dosis bajas no suprimen el NMOR. Es muy probable que el efecto del GABA sobre el sueño sea indirecto, a través de los otros neurotransmisores que tienen una actividad más específica.
Melatonina
La melatonina es la principal hormona de la glándula pineal. Su precursor primario es la serotonina, cuya concentración en la glándula pineal durante el periodo luminoso es superior a la de cualquier estructura del SNC. El nivel máximo de actividad de sus enzimas sintéticas se alcanza durante la oscuridad, por lo tanto el periodo de mayor secreción es por la noche. Es decir, la secreción de melatonina desde la glándula pineal queda inhibida por la luz brillante, por lo tanto la menor concentración de melatonina sérica se observa durante el día.
Se informa que la melatonina aumenta el sueño delta; la administración de 50mg intravenosa acorta la latencia hasta el sueño, mientras que 80mg por vía oral aumenta la somnolencia. A dosis menores se refiere un aumento de latencia a NMOR. La privación de sueño demuestra un aumento de la amplitud de secreción de melatonina, sin cambios de cortisol. La administración contínua de melatonina (2mg) en la tarde originó cansancio excesivo y aumento en la concentración plasmática que excedió entre 10 y 100 veces la concentración fisiológica nocturna.
En el hombre, la melatonina se ha estudiado en relación al síndrome afectivo estacional, conocido como depresión estacional o invernal, donde se refieren bajos niveles de melatonina. Ello se debe a que las terminales nerviosas que activan a los pinealocitos son noradrenérgicas y si en la depresión hay bajo nivel de este NT, explica la baja producción de melatonina Uno de los tratamientos utilizados es el aumento de luz artificial o fototerapia.
Interleucina
Se supone una relación entre el sueño y el sistema inmunitario. En el humano, se ha encontrado que la interleucina 1 (IL-1) sérica se eleva en el sueño, y sabemos que ésta se libera a partir de los macrófagos para activar a los linfocitos T e induce la fiebre por su acción sobre las células hipotalámicas. En animales la IL-1 incrementan el sueño NMOR en forma dependiente de la dosis. También previene el aumento de temperatura cuando se administra por vía intravenosa, lo cual no impide el efecto somnogénico. En el hombre se detecta un pico de IL-1, durante la primera etapa de ondas lentas de inicio del sueño.
LOS SUBTIPOS NEUROQUÍMICOS DE LA DEPRESIÓN
La depresión no posee en ninguno de sus aspectos el carácter de una entidad homogénea. La diversidad de sus causas básicas es el fundamento para considerarla como una agrupación de enfermedades, según hemos reiterado en páginas anteriores. Las causas inmediatas o patogénicas, o sea los mecanismos a través de los cuales las causas básicas determinan el estado depresivo, radican en el plano neuroquímico. Estos factores neuroquímicos distan también de atenerse a una fórmula única. Sobre esta base distinguimos varios subtipos neuroquímicos, como veremos a continuación.
El denominador común de todos los subtipos neuroquímicos radica en la hiponoradrenergia (funcionamiento insuficiente del sistema noradrenérgico). Puede darse hoy por confirmado que cualquier agente estimulante del sistema cerebral noradrenérgico, a través de cualquiera de sus puntos clave (aumento de síntesis de la noradrenalina, inhibición de su degradación, incremento de la sensibilidad o de la densidad de los receptores noradrenérgicos postsinápticos, prolongación del contacto de la noradrenalina con estos receptores), desarrolla una acción antidepresiva, y que toda sustancia inhibidora de la función noradrenérgica hace descender el tono vital y puede producir una depresión por sí misma o con el concurso del desequilibrio de otros sistemas monoaminérgicos, toda vez que el conjunto de estos sistemas funciona a tenor de una interacción recíproca.
La distinción de distintos subgrupos neuroquímicos de depresivos es una tarea que toma una doble base: una base directa, constituida por el estado de los marcadores biológicos y los índices monoaminérgicos y por la modalidad de los psicofármacos antidepresivos más eficaces, y una base indirecta, a tenor de la sintomatología, toda vez que existe cierta correspondencia entre el perfil neuroquímico y la forma psicopatológica. La tipificación de la depresión en el orden neuroquímico es, por tanto, en realidad una tipificación analítica-sintomatológica-psicofarmacológica.
Según nuestra hipótesis de trabajo mantenida en la investigación y en la clínica, por cierto con muy buenos resultados esta última al permitirnos efectuar una selección muy afinada del fármaco más eficaz en cada caso, pueden distinguirse los seis subtipos neuroquímicos de los que yo mismo me he ocupado con amplitud en otros libros y que aquí presentaré en un resumen tan apretado como sistemático.
La depresión hipercolinérgica, definida, como indica su denominación, por la hiperactividad cerebral del sistema colinérgico, constituye la representación genuina de la típica depresión endógena en el plano neuroquímico. De aquí se infieren sus rasgos clínicos: cuadro tetradimensional intenso, con positividad en los marcadores biológicos de depresión más específicos: el acortamiento de la latencia del sueño paradójico, ya descrito al tratar de los trastornos del sueño, y el resultado no supresor en el test de la dexametasona o test Dexa, test que consiste en administrar una pequeña dosis de dexametasona por vía oral para comprobar si suprime o no la elevación de la tasa plasmática de cortisol, y la respuesta selectiva favorable a la terapia con los clásicos fármacos antidepresivos tricíclicos o tetracíclicos, los únicos dotados de acción anticolinérgica.
La depresión hiponoradrenérgica, definida por la actividad insuficiente del sistema noradrenérgico en el cerebro, se distingue de los demás subtipos porque la hiponoradrenergia no se acompaña aquí de otra anomalía neuroquílnica ostensible. Queda incluido en este lugar un amplio sector de las depresiones situativas. Sus manifestaciones en la clínica se extienden por una gama de cuadros diversos. Su índice biológico propio es el descenso en la orina y en el liquido cefalorraquídeo de la tasa del 3-metoxi-4-hidroxi-fenilglicol (MHPG), sustancia producida por la desintegración de la noradrenalina, o sea que es un catabolito suyo, y un descenso plasmático del dihidroxi-feniletilglicol, producto precursor del anterior. Ambas alteraciones no acompañadas por datos anómalos de los catabolitos de otros neurotransmisores. En el plano terapéutico se distingue la depresión hiponoradrenérgica por una espectacular mejoría transitoria con las anfetaminas y una favorable respuesta a los estimulantes noradrenérgicos: la imipramina, la desipramina y la maprotilina entre los productos clásicos, y la mirtazapina, la venlafaxina y la reboxetina entre los recientes.
La depresión hipodopaminérgica, cuya anomalía neuroquímica definidora consiste en una actividad insuficiente del sistema dopaminérgico. Este subgrupo neuroquímico se halla condicionado sobre todo por las causas inmersas en la patología médica, a cuya cabeza se encuentra la enfermedad de Parkinson, como el modelo más demostrativo de hipodopaminergia. El déficit de la transmisión dopaminérgica toma en el Parkinson una extensión doble: afecta a la vez a las vías nigroestriadas, responsables de la sintomatología neurológica, ya la vía mesolímbica, en cuyo lugar se genera la depresión. Se expresa con preferencia por una sintomatología anérgica, de intensidad muy variable, entre el grado ligero y el bloqueo psicomotor. Sus indicadores biológicos más válidos son la hiperprolactinemia y la tasa baja de ácido homovanílico (HVA), el principal catabolito de la dopamina, en el liquido cefalorraquídeo. Es conveniente recordar aquí que existe una correlación positiva entre el nivel humoral de HVA y la actividad psicomotora. La depresión hipodopaminérgica suele mejorar con la administración de un agonista dopaminérgico, tal como la bromocriptina, y sobre todo con los antidepresivos que poseen una cierta acción estimuladora sobre el sistema dopaminérgico, como el bupropion, la nomifensina y el amineptino.
La depresión hiperdopaminérgica, cuyo desequilibrio básico integrado por la concomitancia del exceso de dopamina y la hiponoradrenergia, se debe a la disminución de la enzima dopamínbetahidroxilasa, la enzima responsable de la transformación de la dopamina en noradrenalina. Un dato anómalo en los trastornos bipolares acentuados es el incremento de la densidad de los receptores D2 para la dopamina. Su típica manifestación clínica es la depresión paranoide intensa, a la que se agregan otros cuadros del género de la denominada depresión psicótica. Los marcadores biológicos más específicos suyos son la asociación de una elevada tasa del ácido homovanílico (HVA) en el liquido cefalorraquídeo y una deficiente tasa del 3-metoxi-4-hidroxi-fenilglicol (MHPG) en la orina y en el liquido. Los cuadros hiperdopaminérgicos exigen una terapia mixta, integrada por la combinación de un fármaco antidepresivo pronoradrenérgico con un neuroléptico, cuya administración persigue el propósito de reducir directamente la hiperdopaminergia.
Vayamos ahora con las depresiones vinculadas a los desequilibrios de la serotonina. Entre la depresión hiposerotoninérgica y la hiperserotoninérgica se producen varios cruces sintomatológicos, a causa de que los receptores 5-HT2 que son los receptores de la serotonina subtipo II (la serotonina o hidroxitriptamina se señala con las siglas 5-HT), se apartan del influjo sedativo habitual ejercido por la serotonina en el sistema nervioso, al producir un incremento de la ansiedad, la inquietud psicomotora y la impulsividad, por lo que síntomas de esta clase pueden deberse a la hiposerotoninergia o a la actividad exagerada de los receptores 5-HT2, y análogamente ocurriría con los síntomas inversos (hipoactividad con somnolencia), que podrían estar generados por la hiperserotoninergia global o por el déficit funcional de los receptores 5-HT2.
Hay que permanecer abiertos a la adición de otras observaciones de este estilo, dado el continuo descubrimiento de nuevos receptores serotoninérgicos. La propia sustancia de la serotonina es en sí un tanto problemática, sobre todo en sus relaciones con la depresión, porque si bien ha ganado un merecido crédito como agente antidepresivo, su estructura química es muy semejante a la de la melatonina, que es una hormona de preferente acción depresógena aunque su tasa se halla disminuida en un amplio sector de enfermos depresivos.
La depresión hiposerotoninérgica, cuyo déficit funcional del sistema serotonina puede ser atribuido al elevado nivel de cortisol, originado por el estrés. Sus síntomas se desvían bastante del cuadro depresivo común, en forma de hiperactividad con descontrol: elevada impulsividad, ansiedad, comportamientos de agresividad y actos suicidas, sintomatología originada por el fallo de la acción sedativa ejercida habitualmente por el sistema serotoninérgico sobre la alta actividad corticocerebral. Puede acompañarse de una regulación funcional de los receptores 5-HT2 dominada por la tendencia al incremento. La tasa de triptófano, producto precursor de la serotonina, en el plasma y la del ácido 5-hidroxiindolacético (5-HIAA), el principal catabolito de la serotonina, en el líquido cefalorraquídeo y la orina, se mantienen por debajo del límite normal en este subgrupo de depresivos. La prolactina en el plasma puede hallarse baja al faltar la acción estimulante ejercida por la serotonina sobre su secreción. Su peculiaridad terapéutica más importante es la respuesta favorable a los fármacos agonistas de la serotonina, cuya representación más genuina son los inhibidores de la recaptación de serotonina % que a causa de esta acción prolongan el contacto de la serotonina con el receptor % distribuidos en tres series: los selectivos (no actúan sobre otros neurotransmisores) y globales (actúan sobre todos los receptores de la serotonina) como la fluoxetina, la fluvoxamina, la paroxetina, la sertralina y el citaloprán; los selectivos que no activan el funcionamiento de los receptores 5-HT2 como el trazodone y la nefazodona, que son los más indicados en estos enfermos, y los no selectivos, caracterizados por acompañar la acción proserotoninérgica con una exaltación noradrenérgica, como la venlafaxina y la mirtazapina. Últimamente se ha detectado que los tratamientos farmacológicos contra la hipercolesterolemia (nivel alto de colesterol inducen con cierta frecuencia la presentación de un estado depresivo, en cuya sintomatología son ostensibles la falta de control de los impulsos y las tendencias suicidas, lo cual no ocurre en cambio con el tratamiento higiénico-alimentario del mismo trastorno. Uno de los mecanismos explicativos propuestos para entender la relación causal entre un nivel bajo de colesterol y la aparición de un estado depresivo, es que la reducción del colesterol genera un descenso del número de los receptores de serotonina en el sistema nervioso.
La depresión hiperserotoninérgica muestra una gran afinidad para expresarse por el cuadro de la depresión estacional, cuyo núcleo sintomático está formado por la hipersomnia y la hiperfagia. Los indicadores biológicos suyos más fiables son el exceso de las tasas de triptófano, serotonina y ácido 5-hidroxiindolacético (5-HIAA). En cuanto a los fármacos más eficaces, es curioso que los que ofrecen los mejores resultados sean los activadores noradrenérgicos y los inhibidores selectivos y globales de la recaptación de serotonina (de la serie de la fluoxetina), mientras que los declarados como antiserotoninérgicos, como la mianserina, y los que actúan regulando este sistema al estar dotados por una doble acción contrapuesta, por una parte inhibir la recaptación de la serotonina y por otra disminuir la sensibilidad de los receptores postsinápticos, como la amitriptilina y la nortriptilina, suelen incluso empeorar el cuadro clínico, sobre todo la inactividad y la hipersomnia.
LOS MEDICAMENTOS ANTIDEPRESIVOS
En la familia de los medicamentos o fármacos antidepresivos se incluyen todos aquellos productos químicos que han acreditado poseer la capacidad de reducir total o parcialmente el estado depresivo en un amplio grupo de enfermos depresivos, efecto obtenido la mayor parte de las veces en el plazo de tres a cinco semanas. En las denominadas happy pills (píldoras de la felicidad) en Estados Unidos no se agrupan los productos antidepresivos sino los tranquilizantes menores tipo benzodiazepinas, moléculas desprovistas de acción antidepresiva, capaces incluso de ejercer cierta acción depresógena a partir de la edad de 40 años. Lo más lamentable es que estas sustancias se vienen utilizando algunas veces en el tratamiento de la depresión, lo que implica cuando menos una lamentable pérdida de tiempo o un impulso hacia la encronización del cuadro depresivo. Otra especificación previa necesaria es que la mayor parte de los productos antidepresivos se emplea también en otras indicaciones. Baten el récord en este sentido los productos inhibidores selectivos de la recaptación de la serotonina, utilizados en el tratamiento de estas afecciones: ansiedad/pánico, fobias, síndrome obsesivo, síndrome de estrés postraumático, patología psicosomática, grupo de adicciones sociales y químicas (otro antidepresivo, el bupropion, es muy utilizado en el abandono del tabaco), personalidad límite, déficit de atención por hiperactividad y dolor crónico.
Ante el advenimiento de un amplio lote de medicamentos antidepresivos, se ha venido imponiendo como tarea primordial, con objeto de facilitar su conceptuación y ordenación, la de distribuirlos en grupos. Se ha renunciado a la clasificación a tenor de la estructura química, ya que análogas sustancias ejercen acciones muy diversas y hasta contrapuestas, y en cambio sustancias de lo más dispar pueden coincidir en sus efectos.
Las distintas clasificaciones que han logrado algún predicamento se basan en los siguientes criterios: la ordenación cronológica; el mecanismo de acción farmacodinámico; el efecto neuroquímico, y el efecto sedativo o estimulante. Adelantemos que la clasificación vigente más importante es la que se atiene al efecto neuroquímico.
Con arreglo a la cronología de la aparición del fármaco se distinguen los de la primera generación, que son los antidepresivos antiguos, los de la segunda generación o intermedios y los de la tercera generación, los más recientes. Se trata de una división convencional que sirvió anteriormente para evitar caer en el caos. He aquí la serie de productos incluidos en cada uno de los tres grupos generacionales:
![]()
Antidepresivos de la primera generación: la mayor parte de ellos son los antidepresivos tricíclicos, como la imipramina, desipramina, ciomipramina, amitriptilina y nortriptilina.

Antidepresivos de la segunda generación: un grupo muy heterogéneo por su estructura química, en la que se incluyen entre otros la maprotilina, la mianserina y la viloxacina.

Antidepresivos de la tercera generación: este grupo comprende sobre todo los inhibidores de la recaptación de la serotonina (trazodone, nefazodone, fluoxetina, fluvoxamina, sertralina, paroxetina y citaloprán), más otros agregados también en fechas relativamente recientes como la venlafaxina, la reboxetina y la mirtazapina.
La clasificación basada en el mecanismo de acción del fármaco ha quedado un tanto eclipsada por la que toma como referencia el efecto neuroquímico. Sin embargo, el conocimiento de los distintos mecanismos de acción resulta indispensable para entender cómo se produce la modificación neuroquímica.
El mecanismo de acción representa la vía por la que el fármaco opera para obtener el efecto neuroquímico propio, traducido casi siempre en la activación de un sistema neurotransmisor o de varios. Pues bien, la activación de un sistema neurotransmisor puede obtenerse por alguna de estas vías: el aumento de la síntesis o la liberación del neurotransmisor; la prolongación del contacto de la sustancia neurotransmisora con el receptor postsináptico mediante la inhibición de su recaptación; el aumento de la sensibilidad de los receptores postsinápicos; el aumento de la densidad o número de los receptores postsinápticos; la inhibición de la desintegración del neurotransmisor. Los tres mecanismos sustantivos de los que se valen preferentemente la mayor parte de los psicofármacos antidepresivos son los que modifican la sustancia neurotransmisora, mientras que la acción sobre los receptores postsinápticos ocupa casi siempre un lugar secundario.
Con arreglo al mecanismo operativo quedan los psicofármacos distribuidos en estos tres grupos:

Aumento de la síntesis o de la liberación del neurotransmisor, lo cual puede deberse realmente al incremento de la síntesis a instancia del aumento de la sustancia precursora o del bloqueo o desensibilización del receptor presináptico o receptor inhibidor, llamado así porque inhibe la liberación de la sustancia neurotransmisora a partir de haber alcanzado una cierta concentración en la sinapsis. También se le denomina autorreceptor porque, afincado en el cuerpo de la neurona presináptica y en sus dendritas, ejerce una acción autorreguladora sobre la liberación del neurotransmisor, y receptor somatodendrítico, por razón de su localización (cuerpo de las neuronas y sus prolongaciones dendríticas). La mianserina se distingue por facilitar el incremento de la tasa cerebral de noradrenalina mediante el bloqueo del receptor presináptico y entre los más modernos, la mirtazapina sobresale en este sentido por desarrollar una acción bloqueante sobre los autorreceptores o receptores presinápticos de la noradrenalina y la serotonina provocando el incremento de la liberación de ambos neurotransmisores. Se trata, por tanto, en ambos casos de una acción inhibidora que se traduce en un aumento de la liberación de la sustancia neurotransmisora.
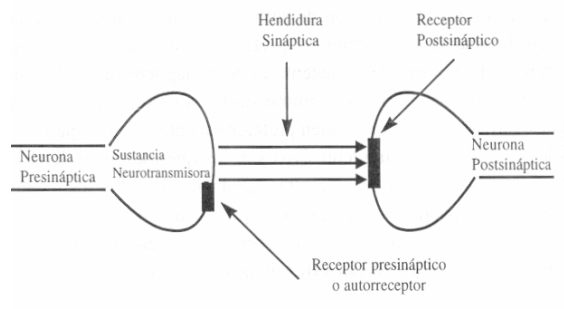
La función de los receptores presinápticos es inhibir la liberación del neurotransmisor en la sinapsis cuando ha alcanzado una concentración suficiente. El bloqueo de este dispositivo autorreceptor constituye el mecanismo de acción de algunos fármacos antidepresivos, que así consiguen incrementar la liberación del neurotransmisor.

La prolongación del contacto de la sustancia neurotransmisora con el receptor postsináptico a causa de la inhibición de su recaptación por parte de la neurona presináptica. Los denominados inhibidores de la recaptación del neurotransmisor son la agrupación de antidepresivos más amplia y eficaz, según veremos después.
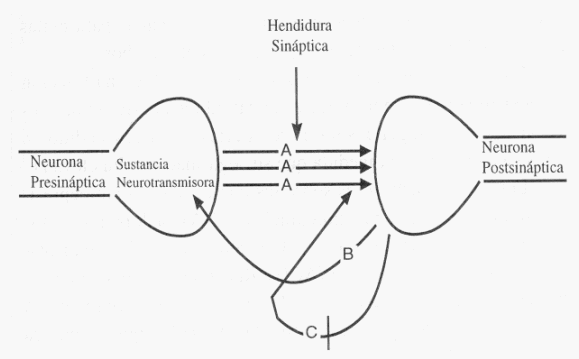
Con la letra A representamos la acción de la sustancia neurotransmisora sobre el receptor postsináptico. La B se refiere a la recaptación del neurotransmisor por la neurona presináptica. con lo que se da fin a su acción sobre el receptor postsináptico. La C indica que la recaptación del neurotransmisor se ha bloqueado con lo que se facilita la prolongación de su contacto con el receptor postsináptico mecanismo de acción propio de los antidepresivos denominados inhibidores de la recaptación de noradrenalina o de serotonina.

La inhibición de la destrucción de neurotransmisor, por lo general a causa de inhibir la actividad de la enzima denominada monoaminooxidasa, que interviene con un papel primordial en la desintegración de los principales neurotransmisores. Se incluyen aquí las sustancias conocidas como IMAO (inhibidores de la monoaminooxidasa)
La utilización de los IMAO se ha mantenido muy restringida porque su efecto clínico es inconstante y difícil de controlar y además porque su administración encierra ciertos riesgos, que únicamente pueden soslayarse evitando su asociación con algunos productos de extenso consumo (queso, habas, vinos, café) y con diversos fármacos antidepresivos. Si no se respeta esta incompatibilidad puede aparecer el "efecto queso", en forma de una crisis aguda de hipertensión arterial con riesgo de muerte, provocada por la acumulación de tiramina. Entre la administración de un IMAO y un antidepresivo de otra familia debe interponerse un intervalo por lo general de dos semanas para evitar que se produzcan interacciones peligrosas entre ambos.
Los nuevos fármacos IMAO actúan sólo sobre una fracción de la enzima MAO, por lo que se hallan libres del riesgo de provocar una crisis hipertensora. Pero la eficacia ha descendido en ellos casi en la misma medida que su seguridad resulta elevada. Los neurotransmisores sobre los que actúan los antidepresivos son la noradrenalina o norepinefrina (NA), la dopamina (DA, precursora de la noradrenalina), la serotonina o 5-hidroxitriptamina (5HT) y la acetilcolina (ACH). Recordemos que la hiperactividad colinérgica se asocia con la depresión endógena y que la administración de agonistas (estimulantes) colinérgicos implica la aparición de sintomatología depresiva. Sobre la acetilcolina ejercen una acción antagónica importante varios antidepresivos clásicos, los denominados antidepresivos tricíclicos, mediante el bloqueo de los receptores postsinápticos colinérgicos muscarínicos. Esta acción representa a la vez un factor terapéutico, que permite a estos productos lograr efectos especialmente eficaces frente a las depresiones más rebeldes, y una desventaja, al ser una fuente de efectos secundarios indeseables (boca seca, visión borrosa, estreñimiento, retención de orina).
Los antidepresivos más utilizados son los que actúan a través de la inhibición de la recaptación de un neurotransmisor, con lo que consiguen prolongar la actuación de este sobre el receptor postsináptico. Por ello, la clasificación que se atiene al efecto neuroquímico se centra en los medicamentos que actúan a través de la inhibición de la recaptación (tabla 6).
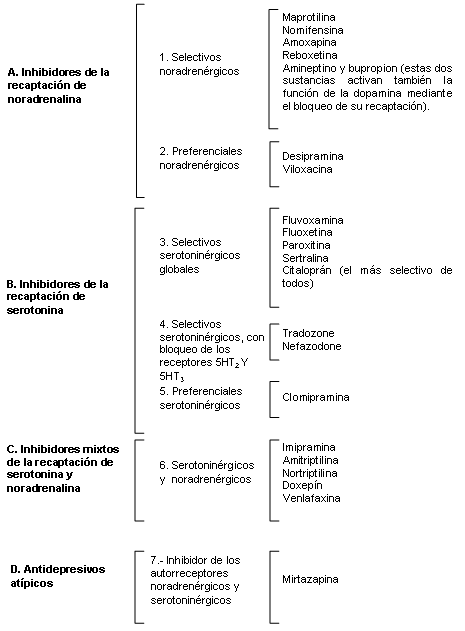
Clasificación de los antidepresivos según el efecto neuroquímico. (Selectivos = acción exclusiva. Preferenciales = acción repartida.)
Por tanto, según nos muestra la Tabla 6, con arreglo al efecto sobre los neurotransmisores se distribuyen los antidepresivos en siete grupos. Precisemos que el antidepresivo atípico por antonomasia, la mirtazapina, un derivado de la mianserina, que ya apuntaba las mismas acciones, es un agonista noradrenérgico y serotoninérgico, cuya acción se verifica sobre ambos sistemas mediante la inhibición de los autorreceptores respectivos, y como al mismo tiempo produce el bloqueo de los receptores serotoninérgicos 5-HT2 y 5-HT3 se agrega aquí la ventaja de evitar los efectos adversos originados por la estimulación de estos receptores: inquietud, irritabilidad, ansiedad, náuseas y disfunción sexual. Debido a su efecto antihistamínico, a causa del bloqueo de los receptores histamínicos H1 o H2, la mirtazapina produce sedación y favorece la instauración del sueño y alguna vez aumento de peso, efecto compartido con otros antidepresivos como el doxepín, la maprotilina y la amitriptilina. De los efectos secundarios indeseables producidos por la estimulación de los receptores 5-HT2 también se libran el trazodone y el nefazodone por ejercer una acción bloqueante sobre estos receptores. Una característica especial del bupropion, a causa de su acción estimulante sobre el sistema dopaminérgico, es acompañarse de una disminución de la tasa plasmática de prolactina.
Con arreglo al efecto propio sedante/estimulante del antidepresivo producido en todos los individuos sean o no enfermos depresivos, se obtiene la escala siguiente:

Sedantes intensos: amitriptilina, trimipramina, doxepín, mianserina, mirtazapina.

Sedantes ligeros: maprotilina, clomipramina, fluvoxamina, paroxetina, citaloprán.

Neutros: imipramina.

Estimulantes ligeros: fluoxetina, sertralina.

Estimulantes intensos: nortriptilina, desipramina, amineptino, bupropion, venlafaxina.
Para la selección del medicamento antidepresivo adecuado para cada enfermo se considera como factor primordial el efecto neuroquímico, puesto que con la administración medicamentosa se trata aquí de corregir el desequilibrio neuroquímico del enfermo, o sea neutralizar la causa última de la enfermedad depresiva. A esta causa última se le llamaba en la Medicina clásica factor patogénico. Por ello, debe catalogarse el tratamiento farmacológico de los enfermos antidepresivos como un tratamiento patogénico y no como un tratamiento etiológico o causal ni tampoco como un tratamiento sintomático. El medicamento actúa como el cable salvador que permite extraer al enfermo depresivo del abismo en que, se halla confinado. Naturalmente, esta acción neuroquímica básica debe complementarse desde el primer momento con una intervención psicosocial, en la que se abordan, dentro de lo posible, los agentes causales fundamentales de la depresión.
La selección de la medicación adecuada para cada enfermo depresivo exige anticipar un juicio sobre la predicción de la respuesta antidepresiva. Para efectuar esta predicción es inexcusable el conocimiento suficientemente amplio y profundo del enfermo, sistematizado en los elementos siguientes: el diagnóstico de la clase de enfermedad depresiva en consonancia con la identidad de sus causas fundamentales; el subtipo neuroquímico, a cuya problemática hemos dedicado un capítulo; el subtipo dimensional distinguido a través de la aplicación del CET-DE; la forma clínica; el estado somático; la edad; la experiencia terapéutica habida en otros posibles episodios anteriores personales y/o familiares.
Sobre la premisa dada por el conjunto de estos datos, la identidad del medicamento adecuado para el enfermo dependerá de estas características suyas: el efecto neuroquímico y el mecanismo de acción, datos que integran su perfil farmacodinámico; el efecto propio sedativo/estimulante; la absorción, distribución y eliminación, o sea las características de su perfil farmacoquinético; y los efectos secundarios. Sobre la ponderación conjunta y equilibrada de los mencionados datos del enfermo y las características citadas de los medicamentos, se hará la reflexión predictiva que culminará en la selección del fármaco antidepresivo considerado idóneo en cada caso.
Con relación a la farmacoquinesia, es preciso especificar que la administración habitual de los medicamentos antidepresivos se realiza por vía oral y su absorción se distribuye entre el estómago y el intestino. Una vez incorporada la sustancia al torrente circulatorio sólo llega al cerebro con capacidad de actuar la fracción del fármaco libre. La otra fracción, ligada a las proteínas plasmáticas, es incapaz de atravesar la barrera hematoencefálica. En los sujetos débiles y en los ancianos, al disponer de un nivel bajo de proteínas en el plasma para fijar una parte del fármaco absorbido, la fracción plasmática activa alcanza mayor nivel, por lo que suele ser suficiente para estas personas el empleo de la mitad o la tercera parte de la dosis habitual del medicamento.
Puesto que con mucha frecuencia se ha de abandonar la monoterapia (terapia por un fármaco) para asociar el antidepresivo seleccionado a otro producto antidepresivo o a un fármaco de familia distinta es conveniente llamar la atención sobre la interacción medicamentosa de tipo farmacoquinético. La mayor parte de los fármacos antidepresivos son potentes inhibidores de alguna de las enzimas del hígado que componen el citocromo P450 (CYP), con lo que vuelven más intensa la acción de aquellos productos metabolizados por la isoenzima inhibida. Por el contrario, otras sustancias como la carbamazepina, producto muy utilizado como timorregulador, inducen la síntesis de enzimas hepáticas, con lo que facilitan la metabolización de algunos medicamentos y en consecuencia disminuyen sus efectos. Conviene estar alerta ante este tipo de interacciones para no dejarnos sorprender por la aparición de unos efectos demasiado fuertes en unos casos y excesivamente ligeros en otros. Puesto que los agonistas serotoninérgicos incrementan la concentración plasmática de los demás fármacos antidepresivos, hay que manejar dosis más ligeras de estos últimos en los tratamientos asociados.
Se ha dado un paso de gigante en la reducción de los efectos secundarios nocivos producidos por las sustancias antidepresivas. Los antidepresivos más antiguos, a causa de su potenciación anticolinérgica, se acompañaban de efectos adversos importantes. La aparición de estos efectos en los tratamientos con los antidepresivos hoy más utilizados, se ha vuelto mucho menos frecuente y de escaso riesgo. A título excepcional, el síndrome serotoninérgico, aunque raro, puede tomar una evolución insidiosa y mortal.
El síndrome serotoninérgico puede complicar el tratamiento con cualquier sustancia de actividad proserotoninérgica y guarda una cierta similitud con el síndrome neuroléptico maligno. Se compone de alteraciones cognitivas (desorientación, confusión, agitación), vegetativas (fiebre, sudoración, diarrea) y neuromusculares (temblores, mioclonias, rigidez, y ataxia o incoordinación). Los cuadros más graves de este tipo son los determinados por la asociación de una sal de litio y un antidepresivo tricíclico o un inhibidor de la recaptación de la serotonina con un IMAO clásico, asociación hoy proscrita precisamente por tal motivo.
Ante un cuadro de hiperserotoninergia se impone la suspensión inmediata del producto serotoninérgico, medida que suele ser suficiente para determinar la rápida desaparición de la sintomatología. En las formas graves del síndrome serotoninérgico es preciso ocuparse además de la deshidratación, la defensa contra la hipertermia y la sedación, y en casos excepcionales echar mano de productos antagonistas específicos de los receptores serotoninérgicos como la ciproheptadina y la metisergida.
Los inhibidores de la recaptación de la serotonina pueden producir también efectos secundarios sobre la esfera sexual. Este trastorno puede ser aliviado o suprimido recurriendo a la disminución de la dosis o a la administración de alguno de estos productos: yohimbina, ciproheptadina, amantadina, buspirona, bupropion.
Los antidepresivos actuales no ejercen una toxicidad sobre los elementos sanguíneos. De esta afirmación taxativa y rotunda se exceptúa la nomifensina, que puede provocar anemia hemolítica, y la mianserina, responsable de algún cuadro de agranulocitosis.
El síndrome de supresión de los antidepresivos, recogido con frecuencia en la literatura en castellano con el anglicismo síndrome de «discontinuación», se manifiesta por síntomas psíquicos, sobre todo ansiedad, irritabilidad, inquietud, y somáticos, sobre todo vértigos, cefaleas, vómitos e insomnio o somnolencia. Aparece entre uno y tres días después de la supresión del medicamento y no se mantiene más allá de dos semanas. Se debe al déficit de serotonina. Es más intenso y frecuente con los medicamentos de vida media breve, como la paroxetina, la fluvoxamina y la venlafaxina, que con los de vida media larga, como la fluoxetina y la sertralina. Pueden evitarse estos síntomas casi siempre mediante una suspensión escalonada. Una buena pauta es reducir la dosis en una cuarta parte cada tres meses. El cuadro desaparece en 24 horas con la administración de algún producto similar o la reanudación del mismo.
En los antidepresivos tricíclicos el síndrome de retirada se atribuye a la reacción de sobreactividad colinérgica y suele manifestarse por síntomas digestivos (náuseas, vómitos, diarrea) y psíquicos (ansiedad, inquietud, hipomanía). Con la brusca suspensión de la clomipramina, cabe la presentación de ambos cuadros (el de la hiposerotoninergia y el de la hipercolinergia).
Hay aproximadamente un 15 por ciento de estados depresivos denominados depresiones refractarias o farmacorresistentes a causa de no ofrecer una respuesta positiva a la administración de una dosis suficiente de la medicación antidepresiva seleccionada. Contra las depresiones refractarias se dispone de estos recursos terapéuticos:
1. La asociación de dos productos antidepresivos o más, con vistas a lograr un refuerzo complementario o sinérgico (el efecto global es superior a la sumatoria de sus efectos individuales).
2. La combinación de la medicación antidepresiva con otras sustancias: un producto psicoestimulante (metilfenidato, dextroanfetamina); el carbonato de litio, sobre todo en las depresiones bipolares; un neuroléptico sedativo (clorpromazina o levomepromazina, haloperidol, etc.) en las depresiones paranoides; una benzodiazepina de vida larga, en las depresiones neuróticas; una sustancia de acción proserotoninérgica como la buspirona (aumento de sensibilidad de los receptores postsinápticos) o el pindolol (bloqueo del autorreceptor presináptico).
3. La agregación de alguna sustancia hormonal: la hormona femenina tipo estrógeno o el principio tiroideo triiodotironina.
4. La potenciación de un mayor despliegue de la psicoterapia o de la intervención psicosocial, o la agregación de la terapia de luz o la supresión del sueño.
5. La suspensión de parte de la medicación asociada o de toda ella (sobre todo las benzodiazepinas de vida larga o media) así como el abandono de todo tipo de drogas con inclusión del alcohol y el tabaco, ya que, además de reforzar el estado depresivo, actúan sobre el hígado como inductores enzimáticos acelerando la eliminación de los medicamentos antidepresivos.
La evaluación de la respuesta antidepresiva es una tarea que sólo puede ejercerse con suficiente finura mediante el empleo de una prueba psicométrica tipo escala. La más empleada en el mundo es la conocida como escala de Hamilton. Para la evaluación del estado depresivo con la referencia del modelo tetradimensional, disponemos de la Escala Tetradimensional para la Depresión (ETD). En trabajos realizados con esta prueba hemos podido comprobar cómo subsistían con mucha frecuencia estados depresivos residuales que pasaban inadvertidos a la observación clínica común.
BIBLIOGRAFIA
1. Avendaño, C. (Coord.). Introduccióna la química farmacéutica (3ª reimp.). Interamericana-McGraw-Hill. Madrid, 1996.
2. Azanza, JR. Guía práctica de farmacología del sistema nervioso central 1999 (1999, 2ª edición). Ediciones. Madrid, 1999.
3. Bradford, HF. Fundamentos de Neuroquímica. Editorial Labor, SA. Barcelona, 1998.
4. Escohotado, A. Historia general de las drogas. Editorial Espasa Calpe. Madrid, 1998.
5. Fruton, JS y Simmonds, S. Bioquímica general. (2ª edición). Ediciones Omega, SA. Barcelona, 1961.
6. García-Blanco Oyarzábal, J. (Ed) Química fisiológica (Vols. I y II) (6ª edición). Editorial Saber. Valencia, 1962.
7. Gómez-Jarabo García, G. (Ed.). Farmacología de la conducta: manual básico para psicoterapeutas y clínicos. (2ª edición). Editorial Síntesis, SA. Madrid, 1997.
8. Goodman y Gilman (Eds.). Las bases farmacológicas de la conducta. (9ª edición) (Vols. I y II). McGraw-Hill-Interamericana. México, 1996.
9. Herrera, E.(Eds.). Bioquímica. Aspectos estructurales y vías metabólicas. (Vols. I y II) (2ª edición). Interamericana-McGraw-Hill. Madrid, 1991.
10. Ondarza, RN. Biología Moderna. (8ª edición). Editorial Trillas. México, 1993.
11. Villar Palasi, V y Santos Ruiz, A. Tratado de Bioquímica (Vols. I y II), (3ª edición). Editorial Augusta, SA. Barcelona, 1968.
Descargar
| Enviado por: | Ale |
| Idioma: | castellano |
| País: | México |
Todos los derechos reservados.