Derecho
Ley de medicamentos en Argentina
ECONOMIA POLÍTICA Y ECONOMIA ARGENTINA
Tema: Ley de Medicamentos en la Argentina:
-
Análisis jurídico
-
Impacto económico
-
Problemática: patentes
ÍNDICE
Introducción...........................................................................................................3
Desarrollo
-
Análisis jurídico..........................................................................................7
-
Impacto económico...................................................................................40
-
Patentes.....................................................................................................63
Anexo..................................................................................................................87
Conclusión...........................................................................................................97
Bibliografía..........................................................................................................99
INTRODUCCIÓN
Contexto económico-social
En el período de tiempo comprendido entre 1998 y 2002 nuestro país sufrió una crisis a nivel económico solamente comparable “a la de los países que atravesaron por guerras o catástrofes naturales” según se pudo leer en un artículo periodístico del diario Clarín del día viernes 21 de junio de 2002, el mismo se titulaba “Se perdieron inversiones por $ 40 mil millones desde 1998”. Semejante noticia no hacía más que reflejar la endeble situación experimentada por la Argentina que tuvo su pináculo en diciembre de 2001. En el cuerpo de esa misma publicación se reflejaban datos que describían un escenario de cuasi-catastrófico:
-
“la caída del PBI del 20,1% entre 1998 y 2002 es la más intensa y duradera de la Argentina”
-
“En el 2001, con la profundización de la recesión y la fuerte suba del riesgo-país, hubo una parálisis de las inversiones”
-
“las empresas no sólo no hicieron inversiones nuevas, sino que no repusieron ni repararon los equipos y las plantas de producción existentes”
-
“la economía argentina no sólo no generó nuevos puestos de trabajo sino que destruyó empleos en forma masiva”
Así las cosas el PBI retrocedió a un nivel similar al de 1993 (medido en millones de pesos de ese mismo año).
Analizando casi medio siglo de movimientos económicos a nivel país no se registran caídas de tal magnitud; gráficamente:
Al analizar las causas de esta inusitada caída del PBI encontramos, por ejemplo, que los consumidores contrajeron sus demandas a causa de las serias restricciones de liquidez que se les impuso a través del denominado “corralito financiero” y a la percepción de pérdida del poder adquisitivo de sus salarios.
En adición, los productores y empresas de los distintos sectores encontraron un límite a sus posibilidades de producción debido a que la “cadena de pagos” estaba “cortada” completamente y a que por la gran incertidumbre existente en el mercado cambiario les era difícil determinar sus costos y la incidencia en sus precios de las variaciones que experimentaría el tipo de cambio.
Esta crisis política y económica del año 2001 además de una caída extraordinaria de la capacidad adquisitiva de la población, produjo un índice de pobreza mayor de 50% (con un índice de indigencia de 25%), con la consecuente disminución del acceso global a la salud y del consumo de medicamentos. Un indicador directo de esta crisis en salud fueron las tasas de mortalidad infantil que permanecieron altas como consecuencia del colapso económico, social y sanitario.
En este contexto los precios de los fármacos subieron en promedio un 60%, llegando algunos a un exorbitante aumento del 350%.
A su vez, en las ventas cayeron aproximadamente un 69% en 2002 en relación a 2001 (una cifra cercana a los USD 2.515 millones). Gráficamente:
Reacción gubernamental
En este marco, el día 9 de enero de 2002 se decreta la Emergencia Sanitaria cuya política en el área de los medicamentos perseguía los siguientes objetivos:
-
Lograr una mayor equidad, entendida como que toda persona debe tener la oportunidad de acceder a aquéllos servicios sanitarios y sociales necesarios
para proteger, promover, mantener y/o recuperar la salud. -
Mejorar el acceso de todos a los medicamentos.
-
Aumentar la eficiencia del gasto en medicamentos no sólo del estado sino
también de la población. -
Garantizar la calidad de los fármacos, tanto los que se venden en las
farmacias como los producidos por los laboratorios estatales.
Para tal altos fines el gobierno decide poner en marcha dos agresivas acciones:
Ley de Prescripción de los Medicamentos por su nombre Genérico.
El llamado Programa (Plan) Remediar, en el marco de la promoción de la Atención Primaria de la Salud.
Oficialmente cada una de estas herramientas es descripta de manera global de la siguiente manera:
1) PRESCRIPCIÓN POR NOMBRE GENÉRICO (DCI)
Objetivos Específicos
Promover la competencia por precio.
Mejorar la calidad de la prescripción.
Medidas:
Obligación del prescriptor de consignar el nombre genérico del medicamento.
Derecho del usuario a elegir según su preferencia.
Habilitación al profesional farmacéutico a dispensar la alternativa comercial elegida.
Instrumentos
Decreto 486 / 02, Resolución 326 / 02
Leyes Provinciales
Ley Nacional 25649 / 02
2) PROGRAMA REMEDIAR
Objetivos Específicos:
Garantizar el acceso de medicamentos esenciales a población de mayor vulnerabilidad.
Fortalecer la estrategia de Atención Primaria de la Salud.
Universalizar la cobertura.
Fortalecer la capacidad asistencial de los sistemas de salud públicos provinciales y nacionales.
Promover el uso racional de medicamentos.
Objetivos Metodológicos de la presente investigación:
Dado este contexto, el propósito del presente trabajo es evaluar el desempeño de esta estrategia gubernamental frente a sus objetivos generales y particulares. Esta evaluación tendrá un doble enfoque jurídico y económico.
Dentro de los temas abordados se destacan 4 ejes neurálgicos:
-
Análisis jurídico de la ley 25.649
-
Análisis del Programa (Plan) REMEDIAR
-
Impacto económico de las medidas adoptadas
-
Conflicto surgido en materia de Patentes de medicamentos
ANÁLISIS JURÍDICO
Tratamiento jurídco de los medicamentos en la Argentina
Adentrarnos en el universo de lo atinente al régimen legal de medicamentos no es tarea fácil. Su estudio es arduo y complejo, puesto que trae aparejado el examen de un plexo normativo vasto, en conjunto con políticas de acceso de la población a dichos productos emanadas desde el Gobierno, que tienden a dar relevancia al tema de fondo, y que por ende no pueden omitirse. Más que nunca toma relevancia, la diferencia existente entre los conceptos pertenecientes al campo de la Química y la Farmacología con los propios de la Ciencia Jurídica, de modo que no es posible intercambiar unos y otros, sin caer en errores lógicos y conclusiones menos verdaderas. Por consiguiente, el abordaje de esta temática debe hacerse interdisciplinariamente.
-
Encuadre normativo general
Las principales normas que regulan los medicamentos son:
-
Ley de Medicamentos 16.463, y su pertinente reglamentación.
-
Ley de Confidencialidad 24.766, arts. 4°, 5° y 6°.
-
Ley de Régimen de Farmacias 17.565
-
Ley de Ejercicio de la Medicina 17.732
-
Resolución MS 326/2002
-
Ley de de Promoción de la utilización de Medicamentos por su nombre genérico 25.649, y su correspondiente reglamentación.
-
La ley Nº 16.463 y su reglamentación
La Ley de medicamentos 16.463, promulgada el 04/08/1964, fija los criterios científicos y sanitarios de aprobación y autorización de comercialización de medicamentos, que dicha norma denomina condiciones, a saber: i) Identidad del principio activo (art. 3° ley); ii) Efecto farmacológico determinado (art. 8°); iii) Calidad, cumpliendo las especificaciones de la Farmacopea Nacional Argentina (FNA) o de otros cuerpos internacionalmente reconocidos (art. 3°); iv) Eficacia comprobada (art. 8°); v) Seguridad (SAFETY) (aspecto relacionado con la inocuidad relativa, art. 5° ley); vi) Ventajas científicas, terapéuticas, técnicas o económicas (art. 7°) y vii) Mantenimiento de finalidades terapéuticas útiles acordes con los adelantos científicos (art. 8°).
Recién en el año 1992 todo el sistema de aprobación de medicamentos experimenta un cambio profundo, a partir de la modificación de la reglamentación sustancial y la transformación de la organización de la Administración Nacional en esta materia, mediante la creación del organismo descentralizado denominado Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT), perteneciente a la órbita del Ministerio de Salud.
El nuevo sistema de aprobación de medicamentos se crea a partir del dictado del dec. 150/92, reglamentario de la ley de medicamentos 16.463, que derogó casi en su totalidad al dec. 9763/64.
Dicho régimen sufrió dos modificaciones importantes por los decretos 1890/92 y 177/93, modificatorios del dec. 150/92, que actualmente regula el registro (autorización), elaboración, fraccionamiento, prescripción, expendio, comercialización, exportación e importación de medicamentos.
Básicamente, el sistema normativo estructurado, establece distintos regímenes de aprobación de medicamentos, definidos por los arts. 3°, 4° y 5° del dec. 150/92 (según dec. 177/93), de acuerdo al tipo de actividad (elaboración, importación, etc.), la calificación del principio activo (ya registrado o novedoso, etc.) o al país de procedencia del producto importado (relacionado con las normas y exigencias sanitarias de cada uno).
Sin perjuicio de lo expuesto, el art. 42 de nuestra Constitución Nacional, reafirma la vertebración del sistema de aprobación de medicamentos a partir de los presupuestos exigidos por la ley 16.463.
Ello es así, en tanto la cláusula constitucional establece que los consumidores de bienes, tienen derecho en dicha relación, a la protección de su salud, seguridad y a una información adecuada y veraz.
Por su parte, los requisitos exigidos por la ley, son explicitados por la reglamentación del dec. 150/92 (según dec. 177/93), en cuanto establece: a) la autorización previa y la inscripción del producto en el Registro de Especialidades Medicinales (REM), art. 2° dec. cit.; b) identidad del producto, mediante la declaración de su fórmula definida y verificable (art. 3° inc. a); c) calidad, información sobre los métodos de control y de elaboración del producto y los datos de biodisponibilidad del mismo (art. 3° inc. b); d) acreditación de la eficacia (art. 5° in fine); e) acreditación de la seguridad (inocuidad relativa, art. 5° in fine).
La política sobre acceso a los medicamentos que el gobierno nacional, a través del Ministerio de Salud, viene implementando, en particular, respecto de los llamados medicamentos genéricos que comenzó, entre otras medidas, con la normativa sobre prescripción de medicamentos por su nombre genérico en el ámbito nacional por res. MS 326/2002 prosiguiendo con la ley de promoción de la utilización de medicamentos por su nombre genérico, está desarrollando un sano y republicano debate, que no está exento de apasionamientos e imprecisiones terminológicas que dificultan el esclarecimiento ciudadano y la formación del consenso de todos aquellos actores sociales y económicos involucrados.
Como regla general, el laboratorio que pretende inscribir un producto que sea una novedad, debe aportar ante la ANMAT, la documentación que represente la realización de distintas pruebas científicamente validadas, como así también, publicaciones relacionadas, que demuestren los extremos antes enumerados. Entre las mencionadas pruebas se encuentran: ensayos de toxicidad y farmacológicos en animales; estudios clínico-humanos controlados para demostrar la seguridad y efectividad del fármaco propuesto.
Esta regla tiene excepciones. Uno de los regímenes de aprobación de las especialidades farmacéuticas se sustenta en el concepto jurídico de (similitud) similaridad de especialidades medicinales con otras ya inscriptas, art. 3° parte final (según dec. 177/93), de tal modo que si una de ellas se encuentra ya inscripta en el registro, por disposición de la norma señalada, la otra puede acceder a su registro, aportando su titular solamente la documentación requerida por dicha norma, lo que conlleva un trámite simplificado que exime de la realización de las pruebas antes mencionadas.
-
Ley 25.649 de Promoción de la utilización de Medicamentos por su nombre genérico
La ley indicada, que fuera sancionada en la Cámara de Diputados en el mes de agosto del año 2002, regula la prescripción por nombre genérico y la dispensa, por parte del farmacéutico, de los sustitutos convenientes, articulando un sistema particular, relacionado con los derechos del consumidor de medicamentos.
Por su parte, se establece para el médico, la obligatoriedad de recetar por el nombre genérico, constituyendo una formalidad esencial, bajo pena de nulidad de la prescripción (arts. 2° párr. 1° y 3°).
Correlativamente, del listado de medicamentos autorizados y comercializados al presente, únicamente el farmacéutico está habilitado, a pedido del consumidor, a "sustituir por" o bien, a dispensar, una especialidad medicinal de menor precio, con igual principio activo, concentración, forma farmacéutica y equivalencia de cantidad de unidades. (art. 2°, párr. 2° y 3°).
En la anterior normativa, se hacía referencia al acto de reemplazo, acto que se encontraba facultado a realizar el farmacéutico de acuerdo al art. 11 Dec . 150/92 según res. MS 326/2002.
Por el art. 9° de la ley, la autoridad nacional creará un listado (Vademécum) de Especialidades Medicinales Genéricas o "formas comerciales autorizadas" "en base a su contenido de principio activo" o "monodroga".
La novedad de la norma consiste en que, por primera vez, se establece una definición de Especialidad Medicinal Genérica: especialidad medicinal identificada por el nombre genérico que corresponde a su composición (art. 4°, ley).
Sin perjuicio de lo dicho, una futura normativa, basada en los antecedentes legales internacionales reseñados, debería definir la categoría de especialidad medicinal genérica en su aspecto técnico, sus caracteres esenciales, los requisitos de aprobación y exigencias a las que deberán adecuarse dichas especialidades.
No obstante, los lineamientos internacionales considerados, no pueden ser extrapolados, sin más, a nuestra realidad para definir técnicamente a estos productos, sino que, a nuestro juicio, resultaría conveniente contemplar la realidad de nuestro mercado farmacéutico y la de los consumidores, todo en función de la garantía del efectivo acceso de la población a los medicamentos.
En torno a la reglamentación de la ley N° 25.649 de utilización de medicamentos por su nombre genérico, se lo hace a través del decreto 987/2003, y constituye un importante capítulo en la historia de la regulación del mercado de medicamentos. Pocas veces un instrumento normativo constituyó un punto de inflexión tan acentuado. Podría, considerarse este decreto como una declaración de guerra. Pero se confunde quien considere que en ella el enemigo son los laboratorios productores. La lucha es contra la falta de acceso a los medicamentos y consiste en cambiar las reglas del mercado. Los laboratorios han operado como causa y consecuencia de dichas leyes pero, al menos en la Argentina de los últimos cinco años, ello no siempre les trajo beneficios. De hecho el volumen de unidades vendidas se había retraído en los años anteriores al dictado de la norma en cuestión a casi a la mitad de los valores registrados en 1999.
Como primer punto de análisis, la Política Nacional de Medicamentos establece un nuevo marco regulatorio con reglas fijas y claras que pueden beneficiar a todo el sistema sanitario en su conjunto. Dicha política se plantea en el decreto de necesidad y urgencia Nº 486/2002 que establece la emergencia sanitaria en el territorio nacional, es ratificada a través de la ley de utilización de medicamentos por su nombre genérico (N° 25.649), e instrumentada luego a través de este decreto.
La prioridad absoluta asumida por la Política Nacional de Medicamentos consiste en promover el acceso de la población a los medicamentos. De acuerdo a la experiencia internacional al respecto se han identificado para ello dos ejes. El primero consiste en la regulación del mercado de fármacos y el segundo en la provisión directa de medicamentos para aquellos que no disponen de medios para adquirir los medicamentos en las farmacias. Al establecer de forma explícita la forma en que deben realizarse las prescripciones de medicamentos, el decreto 987 representa un avance en ambos sentidos.
El decreto establece una adecuada división de roles en la promoción del acceso a medicamentos. En primer lugar, los médicos y odontólogos están obligados a prescribir por nombre genérico (artículo 2°). Aunque se preserva la facultad para incluir en la receta también el nombre comercial del medicamento, se ratifica que serán consideradas no válidas las recetas que no cumplan con este requisito (en el artículo 3º). No hay ni en Argentina ni, probablemente, en el mundo antecedentes de medidas tan drásticas. Esto significa que a partir de la publicación del decreto en el Boletín Oficial en Argentina sólo son válidas las recetas emitidas por nombre genérico.
En segundo lugar, el decreto rescata y jerarquiza el rol del profesional farmacéutico. Este es responsable por verificar la validez de la receta y no puede delegar en dependientes no matriculados las obligaciones contempladas en la ley y su reglamento (Artículo 7º). Pero, además, el decreto asume que el acto farmacéutico involucra no sólo el asesoramiento y correcta información al usuario sobre todas las especialidades medicinales que contengan el mismo principio activo o combinación de ellos y sus distintos precios; sino también responsabilidad legal en dicho acto. El farmacéutico debe documentar este acto profesional consignando en la receta, de su puño y letra, la conformidad del adquirente con relación a la información recibida y al medicamento dispensado, individualizando el mismo por su nombre genérico y de marca comercial o del laboratorio, según el caso, seguido de la fecha, firma y sello donde conste su nombre, apellido y matrícula profesional. También deberá dejar constancia de que informó al usuario al respecto si la receta, además de consignar el nombre genérico, insiste en un nombre comercial.
En tercer lugar, el decreto exige un rol muy activo del gobierno. Otorga un papel central a las autoridades regulatorias, en particular la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica -ANMAT-, en primer lugar porque ratifica que esta institución es la responsable del Registro de Especialidades Medicinales que son las únicas que pueden ser comercializadas en las farmacias argentinas. En segundo lugar, porque debe enumerar las especialidades medicinales que el farmacéutico no estará habilitado para reemplazar, debido a sus características de biodisponibilidad y/o estrecho rango terapéutico. Además fortalece el rol del Estado en la vigilancia y fiscalización al establecer que "ante la falta de información sobre los medicamentos y sus precios por principio activo o combinación de ellos los farmacéuticos serán pasibles de las sanciones de la ley N° 24.240" (Artículo 7º).
Las medidas que han demostrado a nivel mundial un mayor impacto sobre el acceso a medicamentos no consisten en la regulación del mercado farmacéutico, sino en substraer a los remedios de su lógica a través de la provisión directa a la población por parte del Estado o de las instituciones de seguros de salud. En la Política Nacional de Medicamentos esa tarea ha sido asignada al Programa Remediar, implementado para garantizar el acceso gratuito a medicamentos ambulatorios para 15 millones de personas en condiciones de pobreza.
Sin embargo, desde la década del noventa los países más avanzados se han esforzado por incrementar la regulación de los mercados de fármacos, garantizando un mayor acceso a dichos productos. De todas las estrategias políticas posibles, la utilización de genéricos ha demostrado ser la más poderosa en todos los sentidos. Fundamentalmente, porque orienta la competencia de los laboratorios productores por precio y calidad. Medidas ensayadas en décadas pasadas como el congelamiento de precios han demostrado tener efectos adversos serios como el estímulo al agio y a los mercados negros. Algo similar ocurrió con los subsidios directos por parte del Estado, que pocas veces demostraron una asignación efectiva y transparente.
En Argentina se dieron excelentes condiciones que viabilizaron la política de utilización de medicamentos por su nombre genérico. En primer lugar, la calidad de los productos es garantizada por la ANMAT que es una institución pionera en la región y de funcionamiento ejemplar. En segundo lugar, la recesión económica sostenida sumada a una regulación insuficiente condujo a todos los actores del sector a una situación insostenible. Al mismo tiempo que las necesidades sanitarias crecían, el consumo disminuyó, las ventas de laboratorios, mayoristas y minoristas se retrajeron y los financiadores se atrasaron en los pagos o recortaron la cobertura. En tales condiciones un cambio en las reglas de juego del mercado no sólo se hacía necesario sino impostergable. En tercer lugar, en Argentina hay muy pocos productos protegidos por patente, de manera que el mercado de genéricos involucra prácticamente a todas las especialidades medicinales disponibles. Por último, el margen de dispersión de los precios era (y continúa siendo) demasiado alto y esto significa que la substitución de productos permite obtener grandes ahorros sin perjudicar el acceso.
Haciendo hincapié en optimizar el régimen actual, se nos ocurre, como primer punto, regular la publicidad médica. La misma induce una demanda que generalmente no es adecuada ni racional y expande innecesariamente los gastos. En Estados Unidos de Norteamérica los gastos en anuncios directos al consumidor de medicamentos de venta bajo receta representan sólo un 15,7%. Sin embargo, si a ese número se agrega el costo de las muestras que se entregan gratuitamente a los médicos, el porcentaje asciende al 32 %. Muchos países han avanzado en dicha regulación limitando la publicidad, o incluso gravándola con impuestos adicionales.
En segundo lugar, para regular de forma más adecuada privilegiando el acceso se pueden incorporar criterios de costo-efectividad en el registro. Esto significa exigir a los laboratorios oferentes que demuestren que el producto que quieren vender no sólo es más eficaz sino que no encarece innecesariamente el tratamiento. Hasta ahora ningún país lo ha hecho para habilitar la comercialización del producto, pero varios están avanzando en su utilización para las compras públicas y la cobertura de la seguridad social.
Además, este criterio fármacoeconómico se puede aplicar hacia futuro (para habilitar el registro de las nuevas especialidades medicinales) o retrospectiva. Esta última involucra una importante purga del mercado, ya que obligaría a retirar del mercado a aquellos productos para los cuales existan alternativas igualmente efectivas pero cuyo tratamiento involucra costos menores.
Otra línea de medidas imprescindibles consiste en fortalecer la farmacovigilancia incluyendo los aspectos que reglamenta el decreto 987. Esto implica la inspección de todas las farmacias del territorio nacional para verificar el correcto cumplimiento del acto farmacéutico. Entre los aspectos a ser examinados merece destacarse la verificación de: a) que el farmacéutico disponga de las alternativas comerciales más económicas de cada genérico y las ofrezca al consumidor, b) que sólo sea un farmacéutico profesional el que realice la dispensa, y c) que sean rechazadas las recetas no emitidas por nombre genérico.
Por último, y atendiendo a nuestro carácter de país federal, fortalecer la regulación implica favorecer el desarrollo de legislación provincial acorde. En este sentido cabe advertir que además de la ley nacional 25.649 prácticamente todas las jurisdicciones han avanzado en el mismo sentido: Buenos Aires (ley 11.405/02), Ciudad Autónoma de Buenos Aires (ley 752/02), Córdoba (ley 9.010/02), Corrientes (decreto 1.449/02), Chaco (ley 5.048/02), Chubut (ley 4.842/02), Formosa (ley 1.381/02), La Pampa (ley 1.243/90), La Rioja (ley 7.269/02), Mendoza (ley 7.037/02), Misiones (ley 3.843/02), Neuquen (ley 2.392/02), San Juan (ley 7.266/02), San Luis (ley 5.306/02), Santa Fe (ley 12.043/02), Santiago del Estero (ley 6.581/02), Tierra del Fuego (ley 560/02), Tucumán (ley 7.230/02). Además, Entre Ríos, Jujuy, Río Negro y Catamarca tienen leyes que se encuentran en Comisión para adherirse a la ley nacional.
En lo que atañe a la determinación de cuán eficaz ha sido la reglamentación de la ley de medicamentos genéricos, debe decirse, que cuando una política asume como prioridad promover el acceso a medicamentos no corresponde examinar su eficacia exclusivamente a través del monitoreo de precios. Si los precios no varían pero las personas compran las alternativas genéricas más económicas el gasto se reduce y el acceso a los medicamentos esenciales se incrementa.
La Comisión Nacional de Programas de Investigación Sanitaria (CONAPRIS), Estudio Multicéntrico que involucra a Adelco, Universidad Maimónides, CIPPEC (Centro de Implementación de Políticas Públicas para la Equidad y el Crecimiento) e IDICSO (Instituto de Investigación en Ciencias Sociales de la Universidad del Salvador) demostró que la utilización del nombre genérico de los medicamentos involucra un ahorro de $660 millones al año. La estimación surge de aplicar al conjunto de los medicamentos prescriptos exclusivamente por nombre genérico (29%) el porcentaje de ahorro registrado cuando existe sustitución de una marca comercial por otra de menor valor (38%). El resultado se combina con el total del gasto en medicamentos vía farmacia en la Argentina, que se estimó en torno a los 6.000 millones de pesos para el año 2002.
La misma investigación, que también indagó la opinión de médicos, farmacéuticos y pacientes, muestra un importante apoyo de los distintos actores a la ley sancionada en septiembre de 2002, que se tradujo en una rápida difusión de la prescripción por nombre genérico. En efecto, de un total de 4.600 medicamentos con receta analizados en todo el país, en un 57,7% de los casos el médico consignó el nombre genérico. Sin embargo, en más de la mitad de estas recetas también se sugiere una marca comercial determinada. Ello provoca un menor porcentaje de sustitución y por tanto de ahorro para los hogares, el cual podría aumentar significativamente en la medida en que aumente el cumplimiento de la legislación por parte de los profesionales.
-
Un factor relevante a hora de la receta de medicamentos: La Propaganda
La modalidad de recetar por la marca se puede explicar de una forma abierta a muchos factores. Uno de esos puede mostrarse si uno se sitúa en la formación del médico que, entre las disciplinas tratadas se encuentra la Farmacología. En esta se estudian las drogas genéricas y no las marcas.
Con el ejercicio profesional comienza la función prescriptiva y la relación con los productos del mercado. Pero, además, aparece un actor, el agente de propaganda médica de los laboratorios.
Si por las razones que fueren, el médico no dispusiera de una bibliografía especializada y de actualización, se informará entonces de la propaganda médica y de las publicaciones de los laboratorios. Estos divulgan los productos por su nombre comercial o marca.
Así se asocia un efecto farmacológico con la marca; asociación que induce la prescripción de los medicamentos por aquel nombre en detrimento del genérico.
Puede deducirse casi directamente de lo postulado que a mayor llegada de la propaganda, destinada al médico, mayor asociación y una probabilidad más alta de ser recetada la marca.
-
Algunas precisiones conceptuales a partir de las normas de la materia
Medicamento: es toda preparación o producto farmacéutico empleado para la prevención, diagnóstico y/o tratamiento de una enfermedad o estado patológico en beneficio de la persona a quien se le administra.
Principio Activo o Droga Farmacéutica: Toda sustancia química o mezcla de sustancias relacionadas, de origen natural o sintético, que poseyendo un efecto farmacológico específico, se emplea en medicina humana.
Nombre Genérico: Denominación de un principio activo o droga farmacéutica o, cuando corresponda, de una asociación o combinación de principios activos a dosis fijas, adoptados por la autoridad sanitaria nacional o, en su defecto, la denominación común internacional de un principio activo recomendada por la Organización Mundial de la Salud.
Listado de Medicamentos Similares o Bioequivalentes: Nómina de los medicamentos utilizados en el país, designados por su nombre genérico, forma farmacéutica y contenido o composición y de las especialidades medicinales registradas en el país, designadas por el nombre aprobado por la autoridad sanitaria nacional, sea esta una marca de fábrica o comercial o el nombre genérico, y que la autoridad sanitaria nacional considera o acepta como de igual acción o eficacia terapéutica en condiciones similares de uso.
Datos Originales: Son todos los registros o copias certificadas de las observaciones originales, hallazgos clínicos u otras actividades en un estudio clínico, necesarios para la reconstrucción y evaluación del estudio. Tal información incluye: Estudios de laboratorios, memoranda, cálculos y documentos, registro de datos en instrumentos automáticos o exactos, copias verificadas, microfichas, etc. También incluye negativos fotográficos, videograbaciones, microfilmaciones o medios magnéticos.
Resultados: Información obtenida luego de haber finalizado el estudio y que debe ser analizada estadísticamente y comparada con otra información propia o ajena. Generalmente los resultados dan origen a nuevas hipótesis.
Eficacia: Es la capacidad de un medicamento y/o especialidad medicinal para corregir una alteración fisiopatológica o disminuir/eliminar una determinada signo/ sintomatología. La eficacia surge de los resultados del estudio clínico controlado. Entonces, la documentación presentada que cumple con este requisito prueba el extremo de la ley 16.463 (eficacia y seguridad) y será pasible de protección.
Estudio Clínico: Es un estudio sistemático, siguiendo en un todo las pautas del método científico en seres humanos voluntarios, sanos o enfermos realizado con medicamentos y/o especialidades medicinales con el objeto de descubrir o verificar los efectos y/o identificar reacciones adversas del producto en investigación y/o estudiar la absorción, distribución, metabolismo (biotransformación) y excreción de los principios activos con el objeto de establecer su eficacia y seguridad.
-
Responsabilidad Civil en función de los medicamentos
Generalidades
Los medicamentos han logrado un rol fundamental en el desarrollo del ser humano, innúmeros progresos en el tratamiento de las enfermedades, al punto tal que hoy no se conciben terapias que no involucren sustancias medicinales. Coetáneamente, semejante explosión produjo una concentración económica que permitió encarar investigaciones de alta complejidad, hoy son los laboratorios los que inician costosas experiencias que han producido exitosos descubrimientos. El fenómeno convocó a la estadística, la matemática, la computación, la ingeniería, la farmacología y otras áreas del conocimiento que se reúnen alrededor del estudio de una nueva segmentación del saber, generándose una conjunción inesperadamente productiva.
Si pensamos en los efectos adversos, señalaremos que el propio sistema está abriendo los caminos para su superación a través de la biotecnología y la aplicación de procedimientos científicos más sanos.
Sin embargo, estos hechos están reñidos con las valoraciones, puesto que la imagen del sistema transita por los peldaños más bajos en la consideración de las sociedades de todo el mundo.
Hay un nuevo escenario dibujado por la producción, por la venta y por la prescripción masiva de medicamentos. Lejos estamos de aquel galeno que preparaba la poción de medicina para cada paciente. Ello genera también un daño cuantificable, estadístico, "visible" para los ojos del ciudadano, y desvinculado cada vez más de la noción súbita del accidente; es habitual.
Hay una diferenciación de campos entre los laboratorios, médicos, farmacéuticos, publicistas, distribuidores, que son los prestadores del servicio y sus consumidores.
Ello conduce a identificar grupalmente a los presuntos autores del perjuicio en aquellos que sirven el medicamento.
Abonan este impacto las políticas de mercado que colisionan con las razones científicas y las omisiones estatales en el debido contralor.
Valor y disvalor, -las significaciones jurídicas se incrustan en esta fractura, sin poder mantener, muchas veces, el equilibrio necesario.
Una primera hipótesis pregunta para qué hablar de una responsabilidad civil si el propio sistema va brindando sus soluciones, si los daños son menores que los beneficios y deben ser asumidos por la sociedad como un costo del desarrollo. El exceso de regulaciones produce un resultado adverso y ahuyenta las iniciativas innovadoras, desapropiando al sistema de su característica más valiosa, cual es el dinamismo. Se admitirían entonces débitos de reparación en situaciones residuales, como cuando hay culpa grave o dolo.
Otra vía resultaría el postular la protección de la víctima aplicando los factores objetivos de atribución y la reparación plena. El riesgo de estas alternativas suele residir en su simplicidad, en no captar las heterogeneidades que presentan los diversos protagonistas del daño y la propia víctima. Muchas veces se entra en contradicción con otras regulaciones del mismo Estado, y en otras se pretende adjudicar a la responsabilidad civil funciones que no tiene.
El problema no es estar a favor o en contra de la actividad, sino regular su funcionamiento. Ello supone afirmar que la industria debe existir, pero deben corregirse sus efectos disvaliosos, estabilizando el sistema, previniendo los daños, la tendencia a causarlos y reparándolos cuando ya se ocasionaron.
Se trata de ver la responsabilidad como uno de los mecanismos relativos al control social de la empresa, lo cual no implica postular su desaparición.
La cuestión reside entonces en identificar a los distintos protagonistas, qué daños producen cada uno, por qué causas, y los instrumentos de prevención y reparación disponibles. Hay cierta simplicidad en la aplicación de un factor de atribución sobre un responsable; una mirada más lejana, nos vislumbra a la responsabilidad como una constelación que se dispersa en busca de mayor equidad, "dando a cada uno lo suyo".
La responsabilidad se perfila entonces como una distribución de costos de conformidad con criterios económicos, morales y de justicia, siendo ello aplicable principalmente a sectores que tienen aptitud para provocar dicho reacomodamiento. Si los laboratorios tienen beneficios pero externalizan sus costos, puesto que los terminan pagando los consumidores con precios altos y daños no compensados, o el presupuesto de salud de una Nación, es posible que el mecanismo compensatorio reequilibre las cosas.
La responsabilidad también orienta la prevención, puesto que deben cargar con parte del resarcimiento aquellos que están en posición de evitar el daño y no lo hacen; ésta es la función preventiva de la culpa. Si el médico, por ejemplo, sabe que puede responder ante la víctima por recetar desaprensivamente, se cuidará de no hacerlo.
Sin embargo, hay situaciones que escapan a ambos criterios. Puede ocurrir que el galeno medique mal porque hay una limitación para ello que ha sido impuesta por el Estado a fin de beneficiar económicamente al consumidor; o puede ser que el medicamento produzca daños, pero los beneficios son mayores como ocurre en muchos supuestos en que la ciencia determina un daño aceptable. Estos efectos disvaliosos suelen ser eximentes de responsabilidad, ya que son soportadas por la sociedad en su conjunto.
Es un hecho incontrastable que los análisis económicos penetran en la responsabilidad civil; para nosotros, ello no significa una alteración axiológica, es decir, volvernos economicistas. Antes bien supone reubicar el tema de la responsabilidad en las relaciones macrosociales, como corresponde en una sociedad donde los autores y los daños se causan y se sufren sectorialmente; para ello hay que utilizar las ciencias que se ocupan de ello.
Sin embargo, así como en la era de la culpa el débito de responsabilidad era una cuestión de justicia conmutativa, hoy lo es de justicia distributiva. Esta se antepone a la economía que debe frenar sus deseos imperialistas.
Responsabilidad del laboratorio
Cuando se indaga la demandabilidad del laboratorio, hacemos referencia a aquel que es el elaborador, al proveedor de tecnología o quien la adquiere, puesto que el que compra la innovación también carga con los riesgos que ella implica.
Del mismo modo pueden existir supuestos de autoría plural, que suceden ante la falta de identificación del elaborador. Esto ocurre en casos en que la distancia temporal entre el perjuicio y la adquisición del producto, impide saber a quién se compró y quién lo produjo.
En nuestro derecho se ha dicho que, con fundamento en la responsabilidad colectiva, podría arribarse a una autoría plural, presumiéndose el nexo causal con todo el grupo de laboratorios que habrían fabricado el fármaco, en proporción a la participación en el mercado.
Ámbito de la responsabilidad
Existencia de contrato
Puede suceder que la responsabilidad surja en el ámbito de un contrato, sea éste expresa o implícitamente celebrado. Este es un supuesto de excepción, que se da cuando el consumidor adquiere la cosa al fabricante; cuando habiendo eslabones en la comercialización pretende el resarcimiento de quien contrató con él, o bien cuando el que reclama es el intermediario.
En dichos casos se ha fundado la responsabilidad en dos tesis disímiles.
La primera, que compartimos, señala la existencia de un deber de seguridad, implícito o expreso. En el plano de las obligaciones interpartes, este débito es accesorio de las dos obligaciones nucleares del convenio celebrado, pero resulta valorativamente relevante en cuanto el contenido de la prestación comprometida es la preservación de la salud.
En el caso de fármacos, es obvio que se persigue la salud y que la seguridad de la otra parte depende de la calidad del producto elaborado; de modo que estamos ante un fin causalizado, una causa fin que integra la trama prestatoria. Esta motivación, que surge de la intención común de las partes, alcanza al efecto preciso de evitar que el consumidor no sufra daños como consecuencia de un producto que tiene justamente el fin de evitarlos; se trata por lo tanto de una obligación de resultado según la doctrina clásica. Nosotros preferimos señalar, superando la distinción, que dicho débito es el poner todos los medios técnicos, humanos y comerciales de que dispone el fabricante a fin de precaver que el producto sea dañoso.
Cuando se habla de daños, debe precisarse que se trata de aquellos que exorbitan el ámbito propio del perjuicio que ocasiona el medicamento como efecto adverso legitimado por el beneficio que produce.
Hay un deudor profesional a quien se le debe exigir el conocimiento de un experto (laboratorio), y hay una pérdida de control sustancial de la seguridad por parte del consumidor para enajenarla en manos de lo previsto por el primero; surge entonces claramente el débito de seguridad con fundamento en el art. 1198 del Cód. Civil.
Otro sector de la doctrina encontró en estos casos un supuesto de vicios redhibitorios. El art. 2176 del Cód. Civil dispone que si el vendedor (laboratorio) conocía o debía conocer, por razón de su oficio o arte, los vicios o defectos ocultos de la cosa vendida y no los manifestó al comprador, tendrá que indemnizar los daños y perjuicios.
Es interesante destacar que este artículo señala un débito de advertencia que surge del oficio; el experto debe prevenir al inexperto, y si no lo hace incurre en responsabilidad.
El deber de reparar supone la existencia de un defecto oculto al tiempo de la adquisición, que haga impropia la cosa para su destino (art. 2164) y que se halla optado por la rescisión (art. 2176), teniendo en cuenta que esta acción prescribe a los tres meses (art. 4041). La víctima debería escoger la rescisión, lo que es altamente improbable que suceda; el laboratorio podría incorporar una cláusula de renuncia de esta responsabilidad (art. 2166); El defecto oculto es una noción que sólo con mucho esfuerzo puede ser aplicada para explicar normativamente los problemas derivados de medicamentos; hay una imposibilidad de reclamar daños extrínsecos y un brevísimo plazo de prescripción. Estas razones han motivado el rechazo de esa fundamentación.
Inexistencia de contrato
Esta situación es la más común: el laboratorio celebra contratos con los distribuidores del medicamento, lo promociona frente al médico y al paciente, pero no alcanza a vincularse expresamente con el consumidor.
Un grupo de autores ha creído necesario aplicar análogamente los parámetros contractuales, siguiendo la tendencia a la inflación obligacional que se produjo, sobre todo, en el derecho francés. Habría entonces una serie de ventas encadenadas que se van aplicando una sobre otras, que terminan entrelazando convencionalmente al elaborador y al consumidor final que es quien adquiere la propiedad definitiva. Sintéticamente se expresa que el fenómeno contemporáneo de la intermediación en la venta de masas, no debería distorsionar la perdurabilidad de los rasgos típicos del negocio, cuya impronta normativa iría contagiando a quienes acceden a intervenir en el tráfico. Así, la promesa de seguridad que originariamente asume el elaborador, se ensancharía en su manto protectorio hasta alcanzar al consumidor final, sin que éste haya tenido nexo alguno con aquél.
También se ha dicho que la oferta de un producto rotulado por el fabricante, que lleva su marca, se dirige a los consumidores, y siendo aceptada por éstos, se produce el perfeccionamiento del contrato. Los intermediarios se transformarían en "servidores del contrato".
Esta tesis ha tenido recepción jurisprudencial y se ha insistido en algunos congresos en los deberes de lealtad y de seguridad que asume el productor farmacéutico de un modo directo frente a los pacientes.
Se ha criticado esta tesis señalando objeciones de orden técnico, tales como la violación del principio de los efectos relativos de los contratos; la imposibilidad de ceder tácitamente un crédito (art. 1454) o bien la posibilidad de que el elaborador presente defensas típicamente contractuales, como la exceptio non adimpleti contractos, que complicarían la reparación.
Todo ello sin perjuicio de admitir que, sustancialmente, coincide la solución con la del art. 1113 del Cód. Civil, puesto que se impone un deber de seguridad de resultado, que supone un factor objetivo de imputación.
Otro sector ubicó la responsabilidad en el ámbito extracontractual.
Hoy es la doctrina mayoritaria, aunque existen diferencias sobre el factor de atribución.
Una opinión sólidamente sostenida, plantea la atribución de culpa como fundamento de la responsabilidad, invocando el art. 1109 del Cód. Civil. Obviamente no es la acepción clásica de esta imputación, sino una remozada, que incorpora presunciones de culpa derivada del vicio o defecto dañoso, aligeramiento de la carga probatoria puesto que bastaría demostrar una "creencia razonable" y no necesariamente un caso minuciosamente evidenciado, y la noción de que cuando hay daños vinculados al producto, las cosas hablan por sí mismas (Res ÿsa loquitur, thinghs speaks hor himself) y se invierte la carga demostrativa.
Se han tonificado las presunciones a un punto tal que hay autores que se han preguntado qué diferencia existe realmente entre este sistema y el de la atribución objetiva.
La doctrina mayoritaria se inclina por aplicar la imputación objetiva por el riesgo creado por las cosas de las cuales se sirve el fabricante (art. 1113).
Dicha tendencia se acentuará de aprobarse el proyecto de reformas al Cód. Civil, porque se incluiría a esta actividad dentro de las peligrosas que contempla el art. 1113 y el 2176 proyectados.
Más allá de estos debates, lo que interesa es determinar qué débitos concretos se imponen al laboratorio. Sobre este punto coinciden en mucho los autores y la jurisprudencia, sin perjuicio de las discrepancias de fundamentos.
Antijuridicidad
La obligación genérica
Como aspecto nuclear se admite que hay una seguridad prometida al consumidor o razonablemente esperada por éste respecto de la inocuidad del producto; ello involucra la aplicación del principio de buena fe y comprende los deberes de lealtad y completividad de la información, sea que la causa fuente sea contractual o no, ya que siempre el factor de atribución es objetivo.
Cuando hay contrato, hay una seguridad prometida; cuando no hay negocio, hay una seguridad esperada por el consumidor, la que resulta legítima por el principio de buena fe.
Tratándose del primer caso, esta atribución de responsabilidad es objetiva, puesto que surge de una obligación determinada de inocuidad del producto, la que sólo permite eximirse demostrando la culpa de la víctima, la de un tercero o el casus. Cuando no hay convención, se aplica el art. 1113, en cuanto a la responsabilidad por riesgo creado.
Esta doctrina, que siendo poco a poco mayoritaria puede tener recepción legislativa, necesita algunas puntualizaciones en cuanto a los alcances precisos del deber mentado.
Las obligaciones específicas
1) Los vicios de diseño
El laboratorio, titular del registro, puede haber diseñado por sí mismo el medicamento o ser licenciatario. En este último caso debe soportar las cargas peligrosas del mismo, conforme al art. 1113 del Cód. Civil.
El diseño consiste en la programación de un producto con finalidades medicinales, que debe ser aprobado por el Estado. Este diseño siempre resulta riesgoso puesto que todos los fármacos lo son debido a que su peligrosidad debe ser analizada en función de varios parámetros: composición interna, efectos adversos previsibles, imprevisibles, dosis, interacción con otros medicamentos.
El diseño encierra por consiguiente un balance de la relación entre el beneficio que producen y el riesgo que implican.
Si el factor de atribución fuera la culpa, habría que analizar si el laboratorio hizo un fármaco irrazonablemente peligroso (imprudencia) o calculando mal sus efectos adversos (negligencia). En el primer caso, el balance es bastante difícil, sobre todo atendiendo a la existencia de una aprobación estatal; si se basa en la culpa, podría defenderse el laboratorio aduciendo la falta de la misma ya que ajustó su conducta a lo exigible por el ordenamiento nacional.
Si el factor de atribución fuera el riesgo creado, entendemos que no cabría aplicarlo diciendo que todos los medicamentos son peligrosos y entonces siempre es posible sustentar una imputación de responsabilidad. Así como en el caso anterior habría una retracción de supuestos dibitorios, en éste se presentaría una hiperresponsabilidad.
Antes bien, pensamos en la posibilidad de un "vicio" de la cosa. En general la doctrina, cuando explica este tema, señala que hay que demostrar el "error de diseño", pero como estamos en su supuesto de imputabilidad objetiva, preferimos prescindir de esa noción cercana a la culpa. Debe entonces existir un elemento calificante del riesgo de las cosas; así ocurre con los automóviles que lo son, pero no generan la responsabilidad del fabricante ante cualquier accidente.
El medicamento se torna peligroso por la existencia de un vicio; veamos entonces en qué consiste.
El elaborador debe tener los conocimientos de un experto (art. 2176, Cód. Civil), lo que supone estar al tanto de todas las noticias científicas de cómo se elabora un medicamento, cuáles son las medidas de seguridad habituales, cuál es el método científico aplicable. Por lo tanto y a tenor del art. 909 del Cód. Civil, se le exigirá un máximo grado de previsión de las consecuencias dañosas del producto que incorpora al mercado, que sean derivación natural y ordinaria del fármaco (art. 906).
Esta previsibilidad es juzgada en abstracto, ya que no se trata de un pronóstico de culpabilidad. Debe estarse entonces a varios parámetros: a) la utilidad del producto; b) los daños que dicho medicamento causa y su relación objetiva con el beneficio; c) la posibilidad objetiva de hacer dicho medicamento más seguro, lo que puede surgir de la comparación con otros medicamentos en el mercado. Aquí entendemos que el juez puede juzgar la previsibilidad causal tomando en cuenta lo que sucede en otros países, sobre todo tratándose de empresas que tienen un acceso inmediato a dichas posibilidades, como ocurre con los laboratorios. Sabido es que, por múltiples causas, los medicamentos en nuestro país son más riesgosos que en otros, por lo que no habría imposibilidad de tomar en cuenta prescripciones de la F. D. A. de los E. U. Si el laboratorio ha realizado estudios locales que, aún no siendo obligatorios, demuestran que hay una justipreciación selectiva del fármaco ajustada a las coordenadas típicas del país, no habrá evidentemente vicio. Este criterio obviamente redundará en beneficios notorios para el desarrollo nacional; d) el seguimiento del método científico para la investigación y presentación del producto.
Nos interesa detenernos en dos aspectos vinculados a esta previsibilidad "in abstracto". El primero refiere a si las consecuencias mediatas resulta previsibles. El fármaco puede haber sido correctamente diseñado pero mal usado, puesto que se produce una farmacocinética o farmacodinámica distinta por la interacción con otros medicamentos, por ejemplo. En estos casos, existe la ergonomía, que como ciencia debe integrarse a los procesos productivos para calcular cómo actuará el hombre, y por lo tanto resultan predictibles una serie de comportamientos del paciente que tienen rango estadístico. Si el mal uso o la automedicación excesiva son comunes, no son eximentes en tanto son consecuencias mediatas previsibles. Ello sin perjuicio de la interrupción del nexo por parte de una actuación del médico o del farmacéutico.
Otro problema es hasta cuándo debe preverse. Los medicamentos pueden ser perfectamente diseñados y seguros al momento de la aprobación, pero luego de un lapso de tiempo presentar algún efecto adverso no previsto al momento de la calificación estatal. Este es el supuesto de la Talidomida, que desencadenó una secuela de daños varios años después de la ingestión por los pacientes y se evidenció en los hijos de éstos.
Aquí, la doctrina se ha dividido.
Un grupo de autores señala que estos efectos son un riesgo inherente al desarrollo de los medicamentos, que deben ser soportados por los titulares de las empresas. Otro sector, en cambio, participa de la noción de que el fabricante no es responsable si aporta la prueba de que la cosa no puede ser considerada como defectuosa en función del desarrollo científico y de la tecnología prevalente en el momento de su puesta en circulación.
Tratándose de supuestos de responsabilidad objetiva, como dijimos, debe estarse a un pronóstico causal; el problema reside entonces en determinar si dicho examen debe hacerse tomando en cuenta los datos existentes al momento de la elaboración o aprobación del medicamento, o incluyendo también datos posteriores, desconocidos en el instante genético. En este último caso, como bien lo señalan Alterini, López Cabana, se involucra la tesis de Rumelin, que ha sido calificada como ultra objetiva, puesto que toma en cuenta circunstancias no conocidas por el autor.
Sobre este aspecto nos parece necesario observar que la doctrina de la causa debe ser despojada de ese aire metafísico que algunos autores le dieron; este presupuesto trasunta la idea que tiene el legislador acerca de hasta dónde es prudente obligar a alguien a responder; es decir, sugiere un tema de política legislativa. Por ello, convenimos con Llambías cuando señala que corresponde en primer lugar determinar el hecho que ha generado el daño, aplicando un pronóstico objetivo puro, como el de Rumelin; pero luego, en un segundo paso, surge una rectificación que ha impuesto el legislador, orientado por el prisma de la justicia.
Del primer paso, surge que el medicamento ha producido el daño por una causa no presente al momento de la elaboración; del segundo surge la rectificación que corresponde hacer en tanto se trata de una causa mediata no previsible. Por lo tanto no hay responsabilidad por los riesgos del desarrollo.
Sin embargo, corresponde agregar a ello que de acuerdo a los avances actuales de la ciencia farmacológica, compete al laboratorio encarar un estudio de fase IV, post marketing, que permita el monitoreo permanente del medicamento. Si teniendo en cuenta un adecuado estudio de farmacovigilancia se hubiera podido determinar o advertir la presencia del vicio, habrá responsabilidad en tanto éste se torna previsible (art. 904).
Observado el vicio, incumbirá al laboratorio retirar el medicamento del mercado o bien adoptar las previsiones para neutralizarlo.
Nos parece que de este análisis surgirá una particularización de los casos más beneficiosa, que no se contemplaría con la aplicación in totum de la tesis de Rumelin.
2) Vicios de fabricación
En este caso, el diseño es adecuado, pero en la elaboración no se cumple con la finalidad perseguida. La ley 16.463 impone al elaborador de medicamentos la responsabilidad por la pureza y legitimidad de los mismos, y una falla en este aspecto constituirá un vicio de la cosa que demandará la aplicación del art. 1113 del Cód. Civil. Sobre los problemas temporales que surjan nos remitimos a lo expresado en el punto anterior.
Para la prueba de la existencia del vicio, rigen mecanismos probatorios de cierta lenidad en favor de la víctima, que ha propiciado la doctrina. Se trata de la inexigibilidad de una prueba minuciosa y excluyente, sino de demostración de una probabilidad razonable de que el vicio ha ya causado el daño; ello se sustenta en el parámetro de "probabilidad" que pretende la ley (art. 906) y en lo dificultoso que sería aportar un testimonio preciso en un ámbito donde la policausalidad funcional es la regla.
3) Falta de advertencias
El deber del fabricante es advertir al consumidor sobre los riesgos que presenta el producto que ofrece; el laboratorio debe, específicamente, señalar por deber legal (ley 16.463) los riesgos.
Los aspectos más complejos se plantean en cuanto al discernimiento del volumen de la información y los medios a utilizar con el destinatario.
El laboratorio debe presentar una información técnica básica para el registro, relacionada con las acciones farmacológicas, los índices de efectividad, dosis, margen de seguridad, iatrogenia, precauciones, advertencias, contraindicaciones, efecto de acumulación de dosis, posibilidad de generar habituaciones o toxicomanías. Esta información debe ser luego traducida para el médico y para el paciente.
El médico recibe la información fundamentalmente a través de la propaganda que realiza el laboratorio, de las revistas científicas, de los catálogos y de los prospectos que cada medicamento contiene. Casi todos estos medios informativos tienen su origen en el elaborador. En este aspecto señalamos que éste debe preocuparse porque el volumen de la información sea intensivo, teniendo en cuenta la "costumbre" que nos señala una deficiente formación farmacológica del médico en nuestro país. Asimismo debe utilizar medios que lleguen al profesional y no ocultar la información.
En el caso del paciente, el médico se interpone entre éste y el elaborador porque es quien prescribe. De todos modos, el laboratorio debe hacer un prospecto con información básica sobre la posología y los efectos adversos principales. La jurisprudencia de otros países ha señalado que ésta no debe ser excesiva de modo que asuste al consumidor y perjudique al laboratorio. Debe buscarse un equilibrio de información clara, efectiva, que contemple aún las conductas probables que asumirá el consumidor argentino (por ejemplo, en automedicación).
Causalidad
Además de lo ya referido, nos interesa resaltar una nota típica que asume la demostración del nexo causal en los supuestos de daños en masa o "tecnológicos".
En estos supuestos, la víctima debe probar el nexo, el contacto entre la cosa y el daño; llevado a cabo se presume la adecuación y el presunto responsable debe demostrar la ruptura.
-
Doctrina
Relaciones entre tecnología y democracia: e-government y hábeas data. Los bancos de datos relativos a la salud y las obras sociales
La extraordinaria difusión de nuevas formas de informatización y el acelerado avance de la tecnología de las telecomunicaciones impregnaron todo el escenario social dándole una nueva dimensión a conceptos tradicionales como el de democracia y ciudadanía, ampliando no sólo su contenido sino su praxis.
Esto hace que hoy se deban ampliar sus definiciones, incluyéndose los efectos del alto impacto que dicha tecnología produce, fundamentalmente desde el aspecto sociopolítico, sobre las relaciones entre la Administración Pública y los administrados y las modalidades de participación de los ciudadanos en los procedimientos de toma de decisiones públicas y en su fiscalización.
En nuestro país el "gobierno electrónico" ya es una realidad en expansión. El e-government, tal su denominación internacional, se define como la utilización por parte del gobierno de las modernas tecnologías de la información y de las telecomunicaciones para la provisión de servicios y transacciones administrativas vía internet a los ciudadanos, con la finalidad de procurar la eficiencia y la transparencia en su gestión y mejorar el acceso de la comunidad al ejercicio de sus derechos y al cumplimiento de sus obligaciones y cargas.
Al decir de la Cdra. María Rodríguez de Ramírez, la implementación del e-government puede optimizar la gestión de los recursos públicos y lograr una mayor participación ciudadana en el control y monitoreo de los actos de gobierno. Ello está dado en virtud de que mejora la calidad de los productos y servicios que debe proporcionar el gobierno y la provisión de otros nuevos, así como el aumento de la participación de la gente en la elección y eventual provisión de los mismos. Por otra parte, logra la atracción a la esfera del gobierno de sectores de la sociedad que se encuentran excluidos.
Nos encontramos con tres escalas en el avance del e-government: una primera fase en la cual sólo se implementa electrónicamente la normativa vigente, esencialmente la más moderna y de mayor utilización. Luego, en una segunda fase, se otorga al ciudadano la posibilidad de realizar tramitaciones vía internet; hasta que en una última instancia, de mayor optimización, se alcanza no sólo a proveer información y servicios sino también la posibilidad de la comunidad de participar activamente en el proceso de toma de decisiones políticas, en su fiscalización y en el control de su implementación.
Esto coadyuva no sólo a lograr una democracia más extendida sino también a generar un compromiso mayor del ciudadano con la "cosa pública" y su participación activa y responsable en la determinación, en forma compartida con la Administración, de cuáles son las prioridades de la comunidad y cuáles las posibles alternativas de su financiamiento.
Según Stefano Rodotá, "La reducción de la discrecionalidad administrativa, y por lo tanto, también del riesgo del arbitrio y de la gestión clientelar, se une a la adopción de procedimientos automatizados para casos como, por ejemplo, los de la liquidación de pensiones, de la asignación de habitaciones, etc. En estos casos alcanzan relevancia no tanto por la transparencia de los procesos de decisión, cuanto más bien el momento de la igualdad entre los ciudadanos, cuyas posiciones vienen liberadas de la eventualidad de comportamientos discriminatorios derivados de elecciones realizadas por los aparatos de la Administración".
En ese orden, debemos citar el reciente decreto PEN. 378/2005 (LA 2005-B-1722) sobre "Gobierno Electrónico", en el cual se determina el Plan Nacional y los Planes Sectoriales de los organismos de la Administración Pública Nacional, en procura de la eficiencia y la transparencia en la gestión de los organismos de control, intentándose lograr la articulación de los distintos niveles: nacional, provincial y municipal, en pos de satisfacer las necesidades de los administrados y optimizar los recursos públicos disponibles para ello.
En virtud de lo expuesto, se observa que el Estado es el principal "proveedor/productor/tomador" de información, lo que abarca abundantes datos personales de los particulares, cuestión que trae aparejada el derecho de los ciudadanos tanto a la protección de esos datos en cuanto a su difusión y uso como al flujo de esa información entre otros, entrañándose así el principio de la autodeterminación informativa del sujeto.
A partir de lo expuesto podrían identificarse algunas posibles cuestiones a resolver -entre otras- en la efectiva tutela del derecho a la protección de los datos sensibles relacionados con la salud, que se recolectan en este camino hacia la informatización estatal:
Por un lado, el de las nuevas "bases de datos integradas" y la regulación de su acceso, ya que supuestamente el e-government va a permitir la consulta dinámica de las mismas. Si bien el aludido decreto no las menciona explícitamente, se desprende de la norma, y de la práctica cotidiana a la que asistimos en lo personal en calidad de ciudadanos, que necesariamente las mismas se están conformando (vgr., la base creada con anterioridad por el decreto PEN. 1400/2001 [LA 2001-D-4891]).
Por otro lado, nos encontramos con que las bases que almacenan datos relacionados con la salud necesitarán mayor regulación en razón de la creciente complejidad del tema, ya que no resulta suficiente la ley 25326 (LA 2000-D-4363), pues si bien los incluye en general, no soluciona el vacío legal existente respecto de algunas cuestiones especiales relacionadas con el funcionamiento de este tipo de bases, como ser el consentimiento de la persona en tanto paciente, las reglas de confidencialidad de la información clínica y genética, la identidad biológica y la finalidad en la aplicación del uso y posterior destino de la muestra biológica del paciente de la que el "biobanco" obtuvo los datos.
Sobre el particular, y reflexionando con relación a la posibilidad de agregar un anexo a la ley o, mejor aún, crear una nueva para los bancos de datos de salud, más inclusiva y específica, ya se expuso extensamente la problemática en un trabajo anterior donde se consideró -entre otras cuestiones relativas a tales datos- la urgencia de implementar un "hábeas data genético" como parte de una posible solución .
DATOS SENSIBLES CON RELACIÓN A LA SALUD EN LA LEY 25326 Y OTRAS NORMAS
Con referencia al tema que nos ocupa, el art. 6 del Anexo del Decreto de Gobierno Electrónico citado dice que "Los datos en poder del Estado Nacional sobre personas físicas y jurídicas deberán ser protegidos física y logísticamente para que sean tratados conforme a las disposiciones de la ley 25326, cuidando en especial que sean accedidos por personas u organizaciones no autorizadas".
En ese orden, dicha ley y los decretos PEN. 995/2000 (LA 2000-D-4549) y 1558/2001 (LA 2001-D-4950) intentan regular los archivos, registros, bases o bancos de datos públicos y privados destinados a dar información, desde el enfoque de la protección de los datos personales y sensibles que se almacenan en ellos, tanto de personas de existencia física como ideal, estableciendo así el régimen legal del hábeas data.
La mencionada legislación trata la protección de datos de los bancos en general, y quedan pendientes, como ya se dijo, futuras normas complementarias en cuanto al tema del uso y la aplicación de los datos sensibles de bases mucho más específicas, además de las relativas a datos de salud, como aquellas de utilización en el sistema penal , bancos de sangre, de células madre ("troncales"), o bien las que almacenan los datos del perfil genético y/o el proteómico individual que registran algunas entidades u otras que se van creando a posteriori de la ley , y que la misma no llega a abarcarlos en la complejidad de su temática.
Precisamente es en el área de salud -y el Sistema Nacional del Seguro de Salud- donde finca el objetivo de este trabajo, cuestión en la cual la ley 25326 ha sido bastante escueta, vicio legal que resulta factible atribuir -si se quiere- a que la misma vio la luz gracias a la cuestiones planteadas con base en la problemática que presentaban los bancos privados de datos de provisión de información de tipo financiero para los acreedores, dejando de lado aquellas cuestiones que entrañaban un problema para los derechos humanos en sí mismos.
No se debe olvidar, en todo caso, que la norma surgió en las postrimerías de un período bastante largo de neoliberalismo político económico (fue sancionada y promulgada en el año 2000), tiempo en el que se registró un contundente avance en materia de bancarización y globalización económica, con las gravosas consecuencias por todos conocidas, y de muy débil consolidación, en relación con aquélla, de la progresividad en materia de derechos humanos.
Aclarado este punto, conviene en principio analizar la definición conforme a la ley 25326 de la categoría de datos sensibles, cuáles pueden ser considerados como tales, para luego pasar a observar cuáles lo son específicamente con relación a la salud y, en su caso, qué sucede con ellos y cómo se regulan sus bancos.
El art. 2 ley 25326 establece que se entenderán por "datos sensibles" los datos personales "...que revelen origen racial y étnico, opiniones políticas, convicciones religiosas, filosóficas o morales, afiliación sindical e información referente a la salud o a la vida sexual", enumeración que no puede considerarse taxativa, dada su generalización, y que debe ser aplicada armoniosamente a la luz de todo el orden jurídico en general, en el cual encontramos algunas otras leyes especiales que también determinan la confidencialidad y protección de datos de salud.
Basta con citar para ejemplo la ley 25649 (LA 2002-C-3372), de promoción de prescripción y utilización de medicamentos por su nombre genérico, que en el caso de enfermedades crónicas establece la confidencialidad de los datos. También la ley 23511 (LA 1987-B-1745) del Banco Nacional de Datos Genéticos del Servicio de Inmunología del Hospital Durand y su decreto reglamentario, que prescriben la inviolabilidad de los datos genéticos, o bien la Ley Nacional de Sida 23798 (LA 1990-C-2628) y su decreto reglamentario PEN. 1244/1991 (LA 1991-B-1748), pioneros en el tema, que establecen el método de disociación de datos a efectos de que no se pueda identificar al particular con la información relativa a su enfermedad.
Siguiendo el criterio de la LPDP., no son datos sensibles según el art. 5 inc. 2 ap. c de la ley aquellos que se pueden recolectar sin necesidad de consentimiento del titular del mismo: nombre, documento de identidad, identificación tributaria o previsional, ocupación, fecha de nacimiento y domicilio.
Por el contrario, con relación a los datos sensibles de salud se observa que no están enunciados ni definidos exactamente en el art. 2 , y es el art. 8 el que hace nueva mención al prescribir que "Los establecimientos sanitarios públicos o privados y los profesionales vinculados a las ciencias de la salud pueden recolectar y tratar los datos personales relativos a la salud física o mental de los pacientes que acudan a los mismos o que estén o hubieren estado bajo tratamiento de aquéllos, respetando los principios del secreto profesional".
Lo expuesto en principio deviene abstracto en el área de salud, en razón de que alude a los "establecimientos sanitarios" y "los profesionales" vinculados a las ciencias de la salud, al efecto de "permitirles" recolectar y tratar datos personales, entendiéndose por tal (art. 2 ) a la "información referente a la salud", cuestión por demás inoficiosa, no sólo porque ello resulta lógico, adecuado y estrictamente necesario en beneficio del paciente, sino además porque de lo contrario no podrían cumplir con su función específica y ello colisionaría con el resto de la normativa sobre el particular (vgr., la resolución MSyAS. 648/1986 prescribe la obligación de conservar la historia clínica del paciente durante quince años).
A la vez resulta inconducente la prescripción, ya que deriva a los primeros la aplicación de la normativa en materia del secreto profesional que incumbe a los segundos, situación que constituye también un vicio de la ley, ya que los establecimientos sanitarios en general por sus características difícilmente podrían encuadrarse, en casos de incumplimiento, bajo la órbita de la normativa que regula el ejercicio de las profesiones del área . El peso de la ley recaería sobre los profesionales encargados de los bancos exclusivamente pero no sobre éstos, conforme lo ha establecido en el art. 10 de la norma.
Este artículo establece el deber de secreto profesional que obliga al "responsable [del banco] y a las personas que intervengan en cualquier fase del tratamiento de datos personales", incluyendo hasta después de finalizada la relación con el titular del dato. El inc. 2 releva el deber de secreto por resolución judicial y cuando medien razones fundadas relativas a la seguridad pública, la defensa nacional o la salud pública. Esta conjunción lleva a deducir que el relevo sería sólo por resolución judicial, y ésta es la que debe fundarse avalando las circunstancias descriptas por la norma.
Habría que aclarar, además, que en nuestro país la legislación en materia de ejercicio y secreto profesional no se complementa en todos los casos con la correspondiente colegiación obligatoria, fundamentalmente en las nuevas profesiones auxiliares de la medicina que van surgiendo, lo que implica que se carece de una entidad profesional que impulse nueva legislación al respecto, verifique su cumplimiento y se ocupe de su aplicación disciplinaria, omisión como sucede, por ejemplo, en el caso de los psicólogos o los acompañantes terapéuticos.
Asimismo, la LPDP. resulta incompleta al hacer alusión sólo a "establecimientos sanitarios", lo que deja por fuera a un sinnúmero de bancos de datos de prestadores de servicios en general y demás entidades al cuidado de la salud, que también recolectan y tratan datos personales y sensibles a fin de cumplir su cometido. Basta con citar como por ejemplo los centros de día, las obras sociales, los hogares con alojamiento permanente, las escuelas de educación especial, las empresas de informática con "biobancos", las compañías farmacéuticas, etc., todos casos para los cuales no resulta de aplicación el artículo bajo comentario, en atención a que la normativa de aplicación no los considera "sanitarios", ni tampoco son para uso personal.
Es en estas cuestiones en donde la ley nacional difiere de su antecedente primario, la ley española 15/1999, ya que si bien ésta establece una mayor flexibilidad en el tratamiento de los datos de salud con relación a los otros datos sensibles (políticos, filosóficos, religiosos o sindicales), a los que considera de exclusivo resorte del titular, complementa el tema del secreto profesional añadiendo a "cualquier otra persona sujeta asimismo, a una obligación equivalente de secreto", remitiendo a su vez a la legislación de sanidad específicamente.
Siguiendo a Eduardo Molina Quiroga, hay que destacar que España tiene también una importante regulación del tratamiento de los datos de salud en la Ley General de Sanidad 14/1986, que se integra con lo explicitado supra, por lo que la remisión que hace la ley 15/1999 a la legislación de sanidad específicamente está ampliamente justificada, no siendo tan así la indicada en el art. 8 de la ley argentina.
Comentario aparte merece el procedimiento para que el titular de los datos otorgue su consentimiento y éste sea válido: el art. 5 establece en su acáp. 1 que "El tratamiento de datos personales es ilícito cuando el titular no hubiere prestado su consentimiento libre, expreso e informado, el que deberá constar por escrito, o por otro medio que permita se le equipare, de acuerdo con las circustancias".
Cabe destacar que si bien el art. 34 de la norma otorga la legitimación activa para la acción de protección o de hábeas data a "el afectado, sus tutores o curadores y los sucesores...", son innumerables los casos en materia de salud en los cuales el paciente no se encuentra en condiciones de salud física o mental para actuar y tampoco se halla inhabilitado o incapacitado judicialmente como para tener curador o representante legal.
De lo expuesto surge que nuevamente la ley argentina se apartó de la española, ya que la nuestra no permite al tratarse de una cuestión personalísima, y mientras el titular se encuentre con vida aunque momentáneamente imposibilitado, la sustitución de su voluntad por apoderado o por algún familiar o allegado que pueda, en caso de necesidad, interponer la acción de hábeas data en su favor, tal y como concede en su amplitud el art. 43 CN., por ejemplo, para el hábeas corpus.
En la práctica, y considerando que la necesidad del paciente de proteger su intimidad y su autodeterminación informativa pueden requerir urgencia, al igual que su "consentimiento informado" para la conducta terapéutica a seguir, es oportuno considerar que en circunstancias extremas se podría llegar a aplicar por imperio del principio general de analogía, compartiendo el criterio con el Dr. Ignacio Maglio, el art. 21 ley 24193 (LA 1993-A-27) de Trasplantología; en cuanto a las personas que están autorizadas a donar órganos sustituyendo la voluntad del enfermo, podrían también sustituirla para ejercer la acción de hábeas data, en forma similar a lo establecido en su par española.
Situación similar sucede con relación a la determinación de los datos relacionados con la salud: según el art. 2 ley 25326, el "dato sensible" es toda aquella "...información referente a la salud...", generalización cuya vaguedad imprime la necesidad de análisis. Siguiendo la línea del derecho comparado, debe entenderse que cualquier dato de carácter personal que haga referencia a la salud sólo podrá ser recabado o tratado con el consentimiento expreso del titular, de lo que resultaría ser un "dato sensible de salud".
Teniendo en cuenta la Ley General de Sanidad de España ya citada, es un dato de salud protegido "cualquier información" relacionada con el proceso de salud o los procedimientos terapéuticos a los que sea sometido el paciente, además de la ya clásica confidencialidad de los datos de la historia clínica, en cuanto instrumento en el cual se registra, sea cual fuere el medio, el conjunto de toda la información médica y práctica del paciente, sobre la cual se toman las decisiones de las conductas terapéuticas a seguir en beneficio de la salud del mismo.
EL SISTEMA NACIONAL DEL SEGURO DE SALUD Y LAS OBRAS SOCIALES
Así las cosas, corresponde ahora pasar a definir el Sistema Nacional del Seguro de Salud prescripto y regulado por las leyes 23660 (LA 1989-A-51) y 23661 (LA 1989-A-58). El mismo es una organización integrada por agentes del seguro cuyo objetivo fundamental es el de proveer prestaciones de salud a sus afiliados, las cuales deberán ser "igualitarias, integrales y humanizadas, tendientes a la promoción, protección, recuperación y rehabilitación de la salud".
Esta organización es uno de los subsistemas dentro del Sistema Nacional de la Seguridad Social, que, incluido el Instituto Nacional de Servicios Sociales para Jubilados y Pensionados (PAMI.), es el encargado de brindar los servicios indicados en el párrafo precedente para los trabajadores en actividad, jubilados y pensionados.
Por su parte, el decreto 1400/2001 crea una "base integrada" referida a la Seguridad Social, la cual consta de un "Registro de personas" que contiene la información correspondiente a todos los titulares de los distintos subsistemas que la componen (leyes 19032 [ALJA 1971-A-154], 23660 y 23661 , 24013 [LA 1991-C-2895], 24241 [LA 1993-C-3023], 24557 [LA 1995-C-3104] y 24714 [LA 1996-C-3339]) y, por otro lado, la "Base de Vínculos Familiares", que contiene la información relativa a los vínculos existentes entre dos personas que dé derecho a la percepción de los beneficios contemplados en algunos de los subsistemas mencionados.
En ese orden de ideas, se consideran agentes de la seguridad social a las obras sociales, nacionales, cualquiera sea su naturaleza o denominación, a las obras sociales de otras jurisdicciones y a las demás entidades que adhieran al sistema (art. 2 párr. 2º ley 23661).
Es así que la Superintendencia de Servicios de Salud, como autoridad de aplicación del Sistema Nacional del Seguro de Salud según lo dispuesto por el decreto PEN. 1615/1996 (LA 1996-C-3464), no sólo fiscaliza y controla el cumplimiento de la normativa vigente, sino que también procura establecer marcos de acción que propendan al fortalecimiento de los agentes de salud a fin de que alcancen los objetivos generales de la seguridad social, a favor de sus afiliados.
Las obras sociales que integran el sistema descripto son sujetos de Derecho Público no estatal con el alcance que el Código Civil establece en el inc. 2 del ap. 2 del art. 33, contando con autogestión administrativa sin intervención del Estado, pero sí sujetos a contralor estatal, como ya se dijo, al ser una organización de la seguridad social (agente de salud).
De su naturaleza pública se desprende el fin de interés público perseguido por las obras sociales: la protección de la salud pública y el otorgamiento por ley de prerrogativas de poder público, pues perciben las contribuciones y los aportes de trabajadores y empleadores con los que se financia el sistema, lo que nos lleva casi a la "publificación" del servicio de salud que prestan .
De ello resulta que los agentes brindan prestaciones médico asistenciales, a través de servicios propios o contratados, a todo su padrón de afiliados titulares y al grupo familiar que tienen a su cargo. Para ello cuentan no sólo con la información general de cada uno de ellos, sino también con las condiciones -documentadas- bajo las cuales se encuentran los integrantes de ese grupo con relación al titular (suegro, concubina/o, menor bajo guarda, discapacitado, etc.), conforme al art. 9 ley 23660 y resoluciones complementarias.
Acumulan también información, indirectamente, respecto del sueldo que percibe el titular y/o su cónyuge (en el caso de unificación de aportes), en cuanto la obra social recibe un porcentaje del mismo en aportes y en contribuciones del empleador/es (art. 16 ley 23660), lo que le permite también conocer los datos de éstos y hasta las tareas que desempeña aquél (por ej.: a través del convenio colectivo de trabajo, si pertenece a una obra social sindical, de la actividad o bien de personal de dirección) y/o su grupo familiar a cargo (por ej.: hijo soltero mayor de 21 años y hasta los 25 años que curse estudios regulares oficialmente reconocidos, conf. art. 9 inc. a ley 23660).
A eso se suman las respectivas historias clínicas o declaraciones de salud -en casos de enfermedades crónicas, por ejemplo- de aquellos que requieran prestaciones, agregándose además el detalle de éstas y el nombre del prestador elegido para el servicio, los períodos en los cuales se utilizan algunos de los servicios y toda otra serie de datos pertenecientes a la esfera de la intimidad del titular afiliado y su familia derivados de su atención médico asistencial.
En virtud de ello es que debemos adentrarnos en el estudio de los datos que almacenan las obras sociales en pos del desarrollo de su cometido, para poder definir cuál es la información sobre cada afiliado al sistema que pertenece a la esfera de la "autodeterminación informativa" del mismo y para disponer de la cual se necesita su consentimiento expreso, conforme lo establece el art. 5 Ley de Hábeas Data.
LOS BANCOS DE DATOS DE LAS OBRAS SOCIALES
Como ya se expusiera en el acáp. II, el art. 5 inc. 2 ap. c ley 25326 establece qué datos se pueden recolectar sin necesidad de consentimiento del titular, a saber: nombre, documento de identidad, identificación tributaria o previsional, ocupación, fecha de nacimiento y domicilio, en tanto no determina cuáles son exactamente los datos sensibles en materia de salud que deben tener protección y confidencialidad, en su caso, por lo que se reitera lo dicho en cuanto a que se debe proteger toda información referida a la salud de las personas.
Por su parte, el art. 2 de la citada norma en su acáp. 3 define al banco o base de datos como el conjunto organizado de datos personales que sean objeto de tratamiento o procesamiento electrónico o no, cualquiera que fuere la modalidad de su formación, almacenamiento, organización o acceso.
Lo expuesto nos lleva a la primera deducción relativa a las obras sociales y es que deben inscribir su base de datos en el Registro Nacional de Bases de Datos que lleva la Dirección Nacional de Protección de Datos Personales, en conformidad con el art. 21 ley 25326 y su decreto reglamentario PEN. 1558/2001 (LA 2001-D-4850) y las disposiciones complementarias 2/2003 , 4/2004 y 3 y 4/2005 de dicho ente.
En segundo lugar, cabe observar con relación al resto de la información general que recolectan de parte del beneficiario titular, tal como su estado civil, o los datos de su concubino/a, o de sus hijos a cargo, o aquellos familiares o personas a los que destinan un ostensible trato familiar (art. 9 inc. b ley 23660), y hasta podríamos decir la propia afiliación al agente, que podría considerarse un dato sensible conforme al decreto PEN. 1400/2001 .
Sobre la afiliación, un reciente y por cierto discutible fallo de la C. Nac. Electoral no consideró dato sensible la afiliación a un partido político en virtud de que si bien la ley 25326 en su art. 2 preserva las "opiniones políticas" dentro del ámbito del derecho a la intimidad, no supone ello que esa protección deba extenderse a los registros de afiliación político-partidaria, pues si bien la opinión política es reservada, "...no así la afiliación, pues ésta debe ser necesariamente solicitada al partido, el cual -a su vez- la comunica a la justicia electoral...".
Con ese criterio cabría sostener que la afiliación a un determinado agente de salud, teniendo en cuenta en la actualidad el ejercicio del derecho de opción de cambio de obra social, que al ser un acto voluntario se perfecciona con la admisión por parte de la entidad por la cual se opta, y siendo que el mismo debe ser comunicado a los distintos organismos de la seguridad social en virtud de la recaudación y distribución de los recursos, no sería un dato sensible.
Sin embargo, la doctrina no ha sido pacífica al respecto, entendiendo que si bien afiliarse a una persona de Derecho Público no estatal está dentro del derecho a la privacidad y a la intimidad, no se incluye en la esfera de la autodeterminación informativa cuando de esa afiliación deriva su inclusión en registros de carácter público, lo cual no presupone entonces que deba protegerse ese dato de afiliación.
Debe considerarse distinto el caso de las empresas de medicina prepaga, pues las mismas son entidades privadas, por lo que la afiliación a la misma es un dato que necesariamente está dentro de la autodeterminación informativa: o sea que la persona decide si permite o no que se conozca el dato de su afiliación. Cabe recordar que según Guillermo F. Peyrano"...la garantía de la protección de este tipo de datos no se reduce a los datos `íntimos', sino también a todos aquellos datos aptos para identificar a la persona y para establecer `perfiles' de la misma".
De conformidad con lo expuesto hasta aquí, deviene lógico que será una información estrictamente confidencial también para la obra social en el caso en que el afiliado a la misma lo sea en calidad de beneficiario adherente, pues de ello no deriva su inscripción o su anoticiamiento a ningún registro u organismo de carácter público.
En tercer lugar, resulta necesario agregar que al momento de la afiliación debería hacérsele conocer al beneficiario de una obra social el derecho de acceso a la información reconocido por el art. 14 ley 25326 e inc. 6 del art. 4 , en concordancia con el art. 7 decreto 1400/2001, de modo que pueda conocer los datos personales que figuran en la base de datos del agente, como así también los derechos que posee respecto de los mismos de actualización, rectificación, cancelación, supresión o bien aplicación de métodos de disociación.
Asimismo, debería informarse al mismo que sus datos y los de su grupo familiar serán cedidos a los prestadores a los fines de su cumplimiento obligacional, y respecto de los cuales también conserva los derechos enunciados en el párrafo precedente, debiendo poner en conocimiento inmediato del agente cualquier negativa o conflicto al respecto con el ente prestador del servicio.
En cuarto lugar, cabe destacar que, por supuesto, rige para el agente y todos sus prestadores que cuenten con la base de datos del mismo los principios generales de la ley respecto del tratamiento de los datos de los afiliados, a saber:
a) Principio de pertinencia, en cuanto a que la proporcionalidad y calidad de los datos que se recaben y almacenen sean conformes al fin perseguido por el agente.
b) Principio de finalidad, relacionado con el anterior, respecto del objetivo específico para el cual la información personal es recolectada por la obra social o sus prestadores.
c) Principio de utilización no abusiva, es decir, que los datos objeto de tratamiento no pueden ser utilizados para finalidades distintas o incompatibles con aquellas que motivaron su obtención.
d) Principio de exactitud, en cuanto a que los datos deben ser exactos y actualizarse en su caso y que los datos total o parcialmente inexactos o incompletos deben ser suprimidos y sustituidos, o completados por el responsable del archivo en cuanto se tenga conocimiento de la inexactitud o carácter incompleto de la información de que se trate.
e) Principio de derecho al olvido o limitación en el tiempo, que implica que los datos serán eliminados de la base una vez que se haya cumplido el fin para el que fueron recabados, o sea, la desafiliación del beneficiario.
f) Principio de legalidad, que establece que el procedimiento de recolección de datos no debe ser realizado en forma ilícita o desleal.
g) Principio de seguridad en cuanto al tratamiento, cesión y acceso a los mismos. Si bien el decreto reglamentario establece que la Dirección Nacional de Protección de Datos Personales elaborará e implantará las medidas, prácticas y procedimientos que susciten la confianza en los sistemas de información, así como en sus modalidades de provisión y utilización, ello aún no ha sucedido; se estima igualmente que es un principio de cumplimiento obligatorio.
h) Principio del consentimiento en caso de que el mismo sea necesario para la recolección de ciertos datos de carácter sensible, o bien su cesión.
Por otra parte, conviene agregar que también el listado con el detalle de las prestaciones médico asistenciales que se brindan diariamente a cada uno de los afiliados en particular resulta ser de contenido absolutamente confidencial, por lo que cabría implementarse, por parte de las obras sociales, un sistema de disociación de los datos conforme lo indica el art. 2 in fine LPDP. a efectos de proceder al tratamiento de los datos, ya sea con la finalidad de estudio, planificación de estrategias, programas de prevención, o bien de aprovechamiento de los recursos económicos.
Sentado ello, corresponde agregar finalmente que la ley permite la conservación de los datos, su recolección o su tratamiento cuando medien razones de interés general autorizadas por una ley, o cuando la finalidad de dicha circunstancia sea estadística, contable o científica y los titulares de los datos de salud recabados no puedan ser identificados a través de la implementación de algún método de disociación.
Regresan a consideración de esta Procuración del Tesoro de la Nación las presentes actuaciones por las que tramita un proyecto de decreto aprobando la reglamentación de la Ley de Promoción de la Utilización de Medicamentos por su Nombre Genérico 25649 .
ANTECEDENTES
1. Las actuaciones se iniciaron con la incorporación del texto de diversas normas:
a) De la ley 25649, de Promoción de la Utilización de Medicamentos por su Nombre Genérico (v. fs. 1/5).
b) De la ley 24240 , de Defensa del Consumidor (v. fs. 6/9).
c) De la ley 17565 , Reguladora de la Actividad Farmacéutica (v. fs. 10/20).
d) De la ley 17132 , que contiene las Normas para el Ejercicio de la Medicina, Odontología y Actividades de Colaboración (v. fs. 21/43).
e) Del decreto 150/92 , referido al registro, elaboración, fraccionamiento, prescripción, expendio, comercialización, exportación e importación de medicamentos (v. fs. 44/55).
f) Del decreto 341/92 ., por el que se unificaron las sanciones pecuniarias a aplicar por infracciones cometidas contra determinadas normas sanitarias ( v. fs. 56/12).
g) De la Resolución Conjunta ME y OSP 470/92 y MS y AS 268/92 reglamentaria del decreto 150/92 (v. fs. 63/78).
h) Del Formulario Terapéutico Nacional, aprobado por Resolución MS y AS 750/85 (v. fs.79/85).
i) Del decreto de necesidad y urgencia 486/02 por el que se declaró la Emergencia Sanitaria Nacional hasta el 31 de diciembre de 2002 (v. fs. 86/109).
j) De la Resolución MS 326/02 sobre prescripciones médicas u odontológicas por el nombre genérico de medicamentos ( v. fs. 110/116).
k) De la Resolución SP 201/02 por la que se aprobó el Programa Médico Obligatorio de Emergencia (v. fs. 117/125).
2. A fojas 126 la Dirección de Despacho de la Subsecretaria de Coordinación del Ministerio de Salud remitió a la Dirección General de Asuntos Jurídicos de esa jurisdicción una primera versión del proyecto de decreto en consideración.
Dicho servicio jurídico no tuvo objeciones legales que formular (v. 127/128).
3. Seguidamente se remitieron las actuaciones a esta Procuración del Tesoro la que, como recaudo previo, solicitó la opinión de la Dirección General de Asuntos Jurídicos de la Secretaria de la Asuntos Legales de la Secretaría Legal y Técnica de la Presidencia de la Nación (v. fs. 130).
Previa intervención de la Dirección General de Despacho y Decretos de esa Subsecretaria Técnica, que formuló diversas observaciones de carácter técnico-legal (fs.131), tomó intervención el citado servicio jurídico que devolvió el proyecto con otras observaciones (v. fs. 132/149).
4. A fojas 151/164 se incorporó al expediente el proyecto observado.
5. A fojas 166/168 la Dirección General de Asuntos Jurídicos del Ministerio de origen tomó nueva intervención respecto de una reformulación del proyecto anterior, en el que se habrían tomado en cuanta las observaciones formuladas en el área presidencial y que, para entonces, obraría en un anexo no incorporado al expediente. Tras su análisis, concluyó que, respecto de los aspectos legales, no existirían observaciones que formular, opinión que fue reiterada a fs. 170.
6. El Ministro de Salud remitió el nuevo proyecto a la ya referida Secretaria Legal y Técnica señalando que en su elaboración tuvieron en cuenta las observaciones formuladas en esa área, y que el nuevo texto también responde a distintas inquietudes manifestadas por las cámaras de laboratorios, por entidades farmacéuticas y por la Mesa de Diálogo Argentino, lográndose el consenso de los sectores interesados (v. fs.171).
7. El servicio jurídico del área presidencial se expandió nuevamente a las fojas 172/173.
En tal ocasión reiteró lo que ya había señalado en su anterior intervención con el sentido de que Desde el momento que tales sanciones (las previstas en la ley 17132 , referida al ejercicio de la medicina, la odontología y las actividades de colaboración), responden a un determinado tipo penal y atento la naturaleza de las mismas (multa, apercibimiento, inhabilitación, clausura) no corresponde que por vía reglamentaria se haga extensiva su aplicación a supuestos no previstos en la ley. Idéntica observación corresponde efectuar respecto de la aplicación de las sanciones previstas en la ya citada ley 17565 que regula la actividad farmacéutica.
Ello sin perjuicio de citar la opinión del servicio jurídico preopinante en el sentido de que en tanto la ley 25649 habría pasado a integrar un sistema o conjunto normativo junto con las leyes 17132 y 17565 , los incumplimientos en que puedan incurrir los médicos, odontólogos y farmacéuticos, respectos de las obligaciones establecidas por aquella, podrán ser pasibles de las sanciones administrativas que prevén estas últimas conforme a su respectiva profesión.
8. En tal estado esa Subsecretaría solicitó la opinión de esta Casa, previa incorporación del nuevo proyecto que corre agregado a fojas 174/188 y respecto del cual me expediré (v. fs. 188).
LA REGLAMENTACIÓN PROYECTADA
1. Cabe recordar que por la ley 25649 se establece que toda receta o prescripción médica debe efectuarse expresando el nombre genérico o denominación común internacional del medicamento.
Se persigue con ello la defensa del consumidor de medicamentos y drogas farmacéuticas, posibilitando que pueda optarse libremente por las distintas especialidades medicinales existentes en el mercado.
La reglamentación en trámite procura ese objetivo compatibilizando tal derecho con el propio del médico u odontólogo a la libre prescripción.
2. Por art. 1 del Decreto en trámite se aprueba la Reglamentación de la referida ley que, como anexo, integra la norma.
Por el art. 2 se establece que la reglamentación entrará en vigencia a partir de su publicación en el Boletín Oficial.
El art. 3 faculta al Ministerio de Salud para dictar las normas complementarias interpretativas y aclaratorias que fueren menester para su aplicación.
3. Las principales disposiciones de la reglamentación contenida en el anexo, son las siguientes :
3.1. Por la reglamentación al art. 2 de la ley se reafirma la obligación de efectuar toda receta o prescripción médica u odontológica por el nombre genérico o denominación común internacional del principio activo o combinación de ellos, seguido de la forma farmacéutica, cantidad de unidades por envase y concentración, facultando a los médicos y odontólogos a expresar en la receta el nombre comercial o de marca del medicamento, a continuación del genérico.
También se refiere a la obligación del farmacéutico de asesorar al adquiriente sobre las especialidades medicinales que contengan el mismo principio activo o combinación de ellos, y sus distintos precios.
Cuando en la receta se consigne exclusivamente el nombre genérico o este nombre seguido por el de la marca y no se justificase la selección de ella, el farmacéutico debe dispensar, a pedido del consumidor, el medicamento con el mismo principio activo o combinación de ellos, la misma cantidad de unidades por envase, igual concentración y menor precio, anotando en la receta la conformidad del adquirente con la información recibida y con el medicamento dispensado y los datos de este ultimo.
Los medicamentos que el farmacéutico ofrezca en cumplimiento del artículo que se reglamenta deben ser especialidades inscriptas en el Registro pertinente a la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT), elaboradas o importadas por establecimientos autorizados.
Cuando el profesional considere indispensable prescribir por marca, deberá consignar primero el nombre genérico y agregar luego, de su puño y letra la justificación que avale tal decisión con su firma y sello aclaratorio.
En tales casos, si el adquirente solicitase otro medicamento, el farmacéutico deberá informarle que el médico u odontólogo justifico la prescripción por marca. De la decisión del adquirente el farmacéutico deberá dejar constancia en la receta con la firma del interesado seguida de la suya y de su sello aclaratorio.
Si la receta omitiese la cantidad de unidades por envase, el farmacéutico podrá entregar el medicamento que reúna las demás condiciones y que contenga el menor número de unidades.
3.2. Conforme al art. 3 de la reglamentación proyectada establecen A efectos de documentar el correcto cumplimiento de la ley que se reglamenta en todos los casos y cualquiera fuera la modalidad de prescripción, los farmacéuticos deberán registrar en un libro especial rubricado y foliado por la autoridad sanitaria competente los datos que correspondan al profesional prescribiente, fecha de prescripción y de presentación en la farmacia y demás datos del medicamento prescripto y dispensado, así como el número de lote. También corresponderá agregar el nombre de marca y sino (sic) contara con el mismo, el nombre de laboratorio elaborador o importador autorizado por Administración Nacional de Alimentos, Medicamentos y Tecnología (ANMAT) a continuación del nombre genérico del medicamento.
Corresponderá dejar asentados en el libro especial los casos en los cuales la receta se tendrá por no prescripta, así como los supuestos en los cuales en la receta no se hubiere indicado la cantidad de unidades por envase a dispensar.
El cuarto párrafo obliga mantener el libro actualizado y a disposición de las autoridades.
El quinto establece que en el caso que el medico u odontólogo incurriere en incumplimiento a la presente Reglamentación, con relación a los requisitos exigidos para la redacción de la receta tales como la omisión del nombre genérico y/o la forma farmacéutica, y/o la concentración, la cantidad de unidades por envase será pasible de las sanciones previstas en la ley 17132 , sus modificatorias y reglamentarias.
3.3. El art. 7 se refiere a la obligación de los establecimientos autorizados para el expendio de medicamentos de brindar al público, sólo a través de profesionales farmacéuticos autorizados, información sobre los medicamentos con el mismo principio activo o combinación de ellos que el prescripto, y sus precios.
Prevé también que en el supuesto que el farmacéutico no brinde la información a que se refiere la norma o incumpla las disposiciones de la reglamentación proyectada, será posible las sanciones previstas en la ley 17565 y sus modificatorias en el decreto 341/92 o, en su caso, de las sanciones de la ley 24240.
Los adquirientes, frente a incumplimientos por parte de los profesionales alcanzados por las normas proyectadas, podrán realizar la denuncia ante la Autoridad Sanitaria Nacional y esta podrá sancionar a quienes presentaron denuncias maliciosas.
3.4 El art. 8 establece que el Ministerio de Salud, como autoridad de aplicación queda facultado para introducir modificaciones al Programa Médico Obligatorio y fija el ámbito de aplicación del nuevo Formulario Terapéutico Nacional que debe aprobar el Ministerio de Salud.
3.5. El art. 10 , finalmente, faculta a dicho ministerio a suscribir convenios con centros universitarios de estudios con el fin de promover acciones tendientes a la actualización, perfeccionamiento, investigación y transferencia del conocimiento de la temática tratada en la norma proyectada.
ANÁLISIS DE LA MEDIDA PROYECTADA
1. En primer término se observa que la nueva redacción no ha atendido la observación formulada por la Dirección General de Asuntos Jurídicos de la Secretaría de Asuntos Legales de la Secretaria Legal y Técnica de la Presidencia de la Nación en el sentido de que las sanciones previstas en las leyes 17132 y 17565 referidas al ejercicio de la Medicina, la Odontología y las Actividades de Colaboración y la actividad farmacéutica, respectivamente, responden a un determinado tipo penal que, por su naturaleza, no corresponde que por vía reglamentaria se haga extensiva su aplicación a supuestos no previstos en la ley.
2. Esta Procuración del Tesoro ya ha señalado antes de ahora la imprudencia de plasmar conductas punibles penalmente por medio de normas administrativas, en mérito a la flagrante trasgresión que ello supone a la garantía del art. 18 de la CN. (v. dictámenes 188:85).
En tal sentido la Corte Sup. ha dicho que es una de las más preciosas garantías consagradas por la Constitución la de que ningún habitante de la Nación ser penado sin juicio previo fundado en ley anterior al hecho del proceso (Fallos 136:200) y también que Toda nuestra organización política y civil reposa en la ley. Los derechos y obligaciones de los habitantes así como las penas de cualquier clase que sean, sólo existen en virtud de sanciones legislativas y el Poder Ejecutivo no puede crearlas ni el Poder judicial aplicarlas si falta la ley que las establezca (Fallos 178:355).
Es por ello que la configuración de un delito, por leve que sea, así como su represión, es materia que hace a la esencia del Poder Legislativo y escapa a la órbita de las facultades ejecutivas (v. dictámenes 188:85).
2.2. Cassagne sostiene que la unidad del derecho represivo y las garantías ínsitas en el Estado de derecho conducen a la aplicabilidad a las contravenciones de los principios propios del derecho penal sustantivo, entre ellos el Principio de Legalidad, que si bien, en su mayor parte, provienen del CPen., su base radica en los preceptos y garantías constitucionales.
2.3. En el mismo orden, dicho autor, expresa que ...emanado de la cláusula constitucional que instituye la garantía de la defensa, dicho principio (Principio de Legalidad) prescribe que toda pena debe fundarse en ley (art. 18 CN.).
Este precepto, continúa dicho autor (...) tiene entre nosotros una tradición que se ha arraigado en una interpretación jurídica constante, formulada a partir de la sanción de la Constitución de 1853. A su vez el citado principio se completa con la cláusula contenida en el art. 19 , según la cual nadie está obligado a hacer lo que la ley no manda ni privado de lo que ella no prohíbe (...). Interesa advertir que por ley ha de entenderse aquí, en mérito a la naturaleza restrictiva que demanda la interpretación de esta materia -en cuanto tiende a tutelar los derechos fundamentales de las personas- solamente la ley material y formal (Casagne, Juan C.; Estudio de Derechos Público p. 84, Ed. Depalma, Buenos Aires, 1995).
2.4. En igual sentido la doctrina española señala que, al delimitar la ley sancionatoria el ámbito de lo lícito y lo ilícito sancionable, y por tanto, las fronteras de la libertad, este efecto capital en la vida social no puede quedar diferido en una simple norma reglamentaria (García de Enterría, Eduardo, Curso de Derecho Administrativo, 4ª ed., t. II, p.175, Ed Civitas, Madrid).
2.5. Concordantemente Marienhoff ha sostenido que las contravenciones de naturaleza penal no pueden ser establecidas por reglamentos, pues las autoridades administrativas carecen de competencia institucional para crear faltas o contravenciones (Marienhoff Miguel; Tratado de Derecho Administrativo, t. IV, p. 584 y ss, n. 1546 , Abeledo-Perrot, 1980).
3. Por la razones apuntadas observo al último párrafo del art. 3 y al sexto del art. 7 de la Reglamentación proyectada, en le medida en que por vía de extensión hacen aplicables a determinadas conductas que no han sido tipificadas por la ley como contravenciones, sanciones previstas por las leyes que se cita, respecto a otras, en abierta violación al referido principio.
4. En el plano formal formulo las siguientes observaciones:
a) En el primer párrafo del art. 3 debería sustituirse ...se tendrán por no escritas... por la expresión ...se tendrán por no prescriptas. Ello así por cuanto es más correcta la utilización del verbo prescribir, tal como se lo hace en el mismo párrafo, dos párrafos más adelante y en el art. 3 de la ley que se reglamenta.
b) En el párrafo siguiente, segundo del art. 3 luego del punto seguido, debería reemplazarse ...y sino contara con el mismo... por ...y si no contara con él.
c) En el tercer párrafo del art. 7 (que comienza a fs. 184 in fine) debería suprimirse la última parte ...si así lo requiriera el destinatario del servicio, ya que le pedido de éste está mencionado poco antes (... a pedido del adquiriente o consumidor).
d) En el párr. 5 (segundo de fs. 185) debería reemplazarse ...el farmacéutico debe verificar que el destinatario del servicio o adquiriente haya comprendido los alcances de la información recibida, previo a hacer firmar en la receta al adquiriente. Por la expresión ...el farmacéutico debe verificar que el adquiriente haya comprendido los alcances de la información recibida, previo a que firme la receta. Ello así pues no es claro quien es el destinatario del servicio al que se alude, y la expresión hacer firmar no es aceptable.
e) Al comienzo de la párr. 7 del mismo artículo (último de fojas 185) también debería suprimirse ...de bienes o servicios... ya que tratándose de la compra de medicamentos no se aprecia con claridad a que servicios se refiere.
f) En el párrafo siguiente (primero de fs. 186) debería suprimirse la expresión ...ante la autoridad de aplicación... ya que al referirse a ella, poco antes y poco después, como la Autoridad Sanitaria Nacional, podrá llevar a pensar que se trata de una autoridad distinta.
5. Lo que precede, sin perjuicio de recordar que esta Procuración del Tesoro tiene dicho que las cuestiones técnicas su ajenas a su competencia específica ya que su fundación asesora está restringida al análisis de las cuestiones de derecho y su aplicación al caso concreto (conf. dictámenes 230:218, 231:135 y 237:252, entre otros).
Por lo expuesto, una vez subsanadas las observaciones formuladas, considero que el proyecto puede seguir su trámite.
-
Jurisprudencia
Primeramente, haremos referencia a dos fallos de la Corte Suprema de Justicia de la Nación relativos a la responsabilidad penal del médico derivada de la prescripción de medicamentos. Estos son los casos "Abelenda" y "Ahuad": en ambos los imputados recetaron y aplicaron per se o a través de personal paramédico un medicamento ("Lisalgil"), que se ordena como calmante, sin que se advirtiera que el mismo contiene una droga "dipirona", derivada de la pirazolona, puesto que la misma no figura por su nombre genérico en diversos catálogos consultados. Como consecuencia de ello se aplica a pacientes alérgicos, provocándoles un shock anafiláctico que desencadena la muerte de la víctima.
La doctrina está conteste en que el factor de atribución de responsabilidad en estos casos reside en la culpa. En palabra de Bueres, el daño se produce como corolario de la actividad científica pura y no de la actuación autónoma, exorbitante de la cosa. Subyace en ello un examen causal sobre el poder material de actuación del sujeto en el supuesto particular. El medicamento forma parte de un programa de prestación diseñado por el galeno; es un elemento de la terapia donde predomina la conducta. Ello es aún más notorio cuando se omiten controles o pruebas como en los casos que comentamos.
Para evaluar la culpa es moneda corriente recurrir a la clasificación entre obligaciones de medios y de resultado. La Corte prescinde de éste prisma de análisis entrando a la consideración de los deberes concretos del médico, en criterio que compartimos. De todos modos, por el camino de las obligaciones premencionadas se llegaría a similar conclusión puesto que en el suministro del medicamento que se desarrolla dentro de la prestación genérica, es de medios y surge entonces un factor de atribución subjetivo. Podría sin embargo encontrarse quien sugiriera que se trata de un resultado preciso, dar un medicamento inocuo, o bien del incumplimiento de la obligación determinada de hacer pruebas, derivándose de ello un factor objetivo de atribución. Por éste camino se llega a una incertidumbre que consideramos evitable.
Se trata entonces de definir los deberes que tiene el facultativo de acuerdo a la naturaleza de la obligación, circunstancias de tiempo, persona y lugar para deducir de ello la culpa obligacional (arts. 512, Cód. Civil).
Estos débitos deben comprenderse entonces en el contexto prestatorio, es decir tomando en cuenta la relación jurídica que se establece entre el médico y el paciente, el deber genérico de poner todos los medios tendientes a la obtención de la curación esperada, los deberes concretos de efectuar un diagnóstico y una terapia de conformidad con los módulos científicos de la época en que se realiza y las disponibilidades técnicas que tiene para llevarla a cabo, los deberes de seguridad implícitos que la doctrina y jurisprudencia han receptado.
En ese marco aparece el medicamento como herramienta para llevar adelante la prestación comprometida y el paciente como acreedor de los deberes premencionados.
Cabe preguntarse cómo se revelan en el caso, los parámetros que provee el art. 512 del Cód. Civil, a los fines de evaluar la intensidad y límites del débito en cuestión.
Se exige al médico el conocimiento de los de su clase, de su especialidad, en la zona y tiempo histórico en que se desenvuelve la prestación. Se fundamenta ello en el modelo abstracto que, desde "el bonus pater familiae" viene inspirando el juicio de culpabilidad y que la jurisprudencia ha definido como el del "buen profesional de la especialidad". En recientes congresos se ha postulado la adopción del criterio del "profesional prudente y diligente de la categoría o clase en el que sea encuadrable el deudor en cada caso concreto".
La idea de "buen profesional" señala un "deber ser" que otorga preeminencia a la práctica aceptable por sobre la práctica común, lo que señalan las normas de procedimiento científico por sobre las claudicaciones de la realidad. Así se resolvió en un caso donde el informe experto señaló que no es habitual realizar pruebas si el paciente no informa de su alergia, diciendo el juez que "el hecho no significa sino que existe una práctica maliciosa, totalmente reñida con las normas de la prudencia y no menos con las propias reglas del arte".
Tanto este módulo abstracto, como su apartamiento en casos concretos, se compadece con las ideas difundidas en el derecho comparado. En el área anglosajona se cae en la culpa cuando la conducta desarrollada es inferior a la que la sociedad considera aceptable; el médico tiene discrecionalidad pero debe inclinarse siempre por un método o práctica aceptado, aunque sea por una minoría respetable de colegas. Pero esta regla ha sido moldeada en la realidad puesto que aceptar reglas locales puede favorecer la creación de islas o focos de prestación por debajo de lo normal y aceptable Las Cortes han dejado de lado el módulo abstracto cuando las prácticas que de él se derivan son irritantes. Para apartarse del modelo se suele tener en cuenta el costo de la prestación exigida y la necesidad de evitar la medicina defensiva; si con una prueba rápida, de bajo costo se hubiera evitado el daño, ésta es exigible aún cuando no sea de práctica hacerlo en la zona o especialidad.
Los deberes del médico con referencia al medicamento no deben ser considerados en general, sino en relación a la terapia elegida.
En primer lugar debe hacer un buen diagnóstico. Debe evitar el tratamiento medicamentoso sobre la base de síntomas; al medicar rápido para poder atender muchos pacientes en la jornada de trabajo. Debe, por el contrario, reavivar la clínica como medio de diagnóstico, dedicar tiempo al paciente, conocer su pasado.
Sobre esta base el galeno puede medicar, puesto que dar un fármaco requiere un manejo adecuado de la relación riesgo/beneficio; es en sentido técnico un acto de experimentación que no puede abordarse sobre la base de un conocimiento breve, meramente sintomático y por lo tanto altamente riesgoso. Es probablemente lo que ha sucedido en los casos fallados.
Lo dicho sirve para interpretar la intensidad del deber genérico del facultativo. En los casos que comentamos había pruebas de que el paciente había advertido que era alérgico, incluso llevaba uno de ellos una medalla indicativa al respecto.
Pero si así no hubiera ocurrido, cabe preguntarse si es deber del médico averiguarlo. Entendemos que una buena práctica hubiera sido el diagnóstico clínico y de el se hubiera desprendido la alergia. En cambio si de un diagnóstico prudente es normal que no se adviertan algunos estados preconstitutivos, éstos serán eximentes.
El galeno no tiene la obligación de conocer las características de todos los medicamentos, sino la del que él elige porque considera adecuado como medio para la curación. Así lo expresa el procurador de la Corte, Andrés D'Alessio, en la causa "Abelenda", al precisar que "El hecho de que exista gran cantidad de productos no lo exime (al médico) de ninguna responsabilidad; por el contrario, según mi criterio, ello crea en cada profesional una obligación aún mayor de conocer qué es lo que receta".
La discrecionalidad técnica y la especialización que tiene el médico fundamenta esa responsabilidad (art. 902, Cód. Civil).
En aquellos supuestos en que la relación médico-paciente es prolongada, la cautividad del paciente aumenta. Suele suceder que el médico prescriba medicamentos durante largos períodos de tiempo, al final de los cuales se presentan secuelas dañosas. Es el caso habitual de los enfermos mentales, depresivos, ancianos; prueba de ello es el alto consumo de tranquilizantes en Argentina.
En el derecho comparado se ha responsabilizado al facultativo por su imprevisión al generar adicción en el paciente. En nuestro sistema jurídico podría argüirse dicha responsabilidad con fundamento en la negligencia profesional al no informar debidamente al paciente sobre los efectos que entraña dicha medicación prolongada, o bien al no justificarla debidamente en un estado de necesidad.
Precisamente, en este tema tiene radical importancia el consentimiento informado del paciente. Recientes congresos nacionales han señalado que es deber del profesional "informar" a su cliente.
IMPACTO ECONÓMICO
Estamos analizando la ley N°25.649, que fuera sancionada el 28 de agosto de 2002, la cual establece que toda receta y/o prescripción médica u odontológica debe efectuarse expresando el nombre genérico del medicamento, seguida de forma farmacéutica, cantidad de unidades por envase y concentración.
Asimismo, el profesional farmacéutico deberá informar al público sobre todas las marcas comerciales que contengan el mismo principio activo, con la misma cantidad de unidades, forma farmacéutica y concentración, y los distintos precios de cada uno de esos productos. De este modo, el consumidor puede elegir la marca y precio del medicamento recetado por el médico. En ningún momento autoriza la sustitución de la droga prescripta por el profesional médico u odontólogo.
Veremos a continuación cual es el impacto económico que trae aparejada la sanción de esta ley.
Para ello debemos analizar en primer lugar la política aplicada por el Gobierno, así advertiremos los fines buscados y compararlos con los resultados obtenidos a nivel económico y social.
En cuanto a la prescripción por nombre genérico los objetivos específicos del Gobierno son los de promover la competencia por precio y mejorar la calidad de la prescripción. Y en cuanto a las medidas adoptadas para el caso están: obligación del prescriptor de consignar el nombre genérico del medicamento; el derecho del usuario a elegir según su preferencia; la habilitación al profesional farmacéutico a dispensar la alternativa comercial elegida. Para ello se adoptaron diversos instrumentos, como ser el Decreto 486 / 02, Resolución 326/02, la aplicación de Leyes Provinciales y la Ley Nacional 25649 / 02 ya mencionada.
No obstante el gobierno adopta otras dos herramientas para afrontar el problema. Cuyos objetivos específicos perseguidos son: garantizar el acceso de medicamentos esenciales a población de mayor vulnerabilidad; fortalecer la estrategia de Atención Primaria de la Salud; universalizar la cobertura; fortalecer la capacidad asistencial de los sistemas de salud públicos provinciales y nacionales; promover el uso racional de medicamentos.
Estas herramientas fueron buscadas a causa de la profunda crisis existente a principios del año 2002, cuando la situación económica, financiera y social de Argentina había alcanzado un nivel de gravedad extremo. El fuerte deterioro de la coyuntura que, claramente, se reflejaba en una pérdida de bienestar de la población, tras la marcada caída del producto bruto, los elevados niveles de pobreza y de desempleo, el desfinanciamiento de todos los agentes del sistema, las dificultades crecientes para afrontar los compromisos asumidos con el exterior, y, como consecuencia de ello, el severo desbalance de las cuentas fiscales, inevitablemente desencadenó una profunda crisis social, que acentuaba, a tasas exponenciales, el surgimiento de conflictos. Argentina había entrado en un círculo vicioso donde la gravedad de tal escenario parecía irreversible; la crisis política era cada vez más preocupante y la ausencia de soluciones inmediatas realimentaba el estado crítico del país.
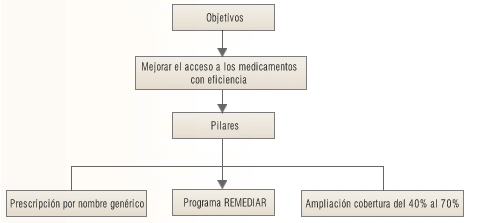
Correlativamente la salud de la población fue una de las áreas más perjudicadas. El sistema de provisión de medicamentos se encontraba en franco deterioro. Los centros asistenciales enfrentaban serios problemas con el suministro de medicamentos e insumos básicos, tras los cambios de precios relativos resultantes de la liberalización del tipo de cambio. La retracción de los ingresos del sistema de seguridad social, originada en la caída significativa de los empleos formales, el incremento de la tasa de desocupación y la baja de los salarios, imposibilitaba el cumplimiento adecuado de las prestaciones obligatorias hacia sus beneficiarios, al tiempo que lo aproximaba al quebranto financiero y al colapso institucional. A su vez, la disminución de la población con cobertura (bajo algún modelo de aseguramiento) se reflejaba en un desplazamiento constante de la demanda de servicios y medicamentos hacia el sector público.
Paralelamente, el empeoramiento de la distribución del ingreso nacional tornaba el gasto en salud cada vez más regresivo, como consecuencia de la caída del poder adquisitivo doméstico, el fuerte encarecimiento de los medicamentos y la menor cantidad de personas con cobertura.
Así, sin poder evitarlo, los efectos negativos de la crisis se habían plasmado en un importante deterioro del acceso de la población a los servicios de salud, y especialmente a los medicamentos, lo cual obligó al gobierno a plantear medidas de política con carácter urgente, que permitieran de un modo u otro paliar los perjuicios que, el cambio fundamental en las condiciones económicas domésticas ocurrido en ese momento, generó sobre la sociedad. Por ello, en el marco de la Ley Nacional 25.561 de Emergencia Pública y Reforma del Régimen Cambiario de diciembre de 2001 que declara la emergencia en materia social, económica, administrativa, financiera y cambiaria a nivel nacional, se establece la Emergencia Sanitaria Nacional, mediante el Decreto Nacional Nº 486 en marzo de 2002, cuyo objetivo fundamental era garantizar a la población argentina el acceso a los bienes y servicios básicos para la conservación de la salud, a partir de:
-
Restablecer el suministro de medicamentos e insumos a las instituciones públicas con servicios de internación
-
Garantizar el suministro de medicamentos para tratamientos ambulatorios a pacientes en condiciones de alta vulnerabilidad social
-
Garantizar el acceso a medicamentos e insumos esenciales para la prevención y el tratamiento de enfermedades infecciosas
-
Asegurar a los beneficiarios del sistema de seguridad social (incluido el sistema de servicios sociales para jubilados y pensionados -PAMI-), el acceso a las prestaciones médicas esenciales.
El cumplimiento de ese conjunto de objetivos implicaba, frente a la crítica situación económica y social, la implementación de medidas que, en el corto plazo, lograran alcanzar un elevado nivel de efectividad y mejorar el bienestar de la población, cuidando al mismo tiempo de no alterar el funcionamiento y la transparencia del sistema de salud en general. Específicamente, el Ministerio de Salud fue facultado a dictar normas complementarias destinadas a implementar los siguientes instrumentos en tres frentes: -
Acceso a los medicamentos para todos
-
Disponibilidad de medicamentos e insumos críticos en centros asistenciales
-
Fortalecer el Sistema de Seguridad Social en el área medicamentos
-
Botiquín de 21 presentaciones: 1.447 Dosis Diarias Definidas (DDD) y 154 tratamientos;
-
Botiquín de 34 presentaciones: 1.882 DDD y 228 tratamientos.
-
¿A quién le compra el Remediar?
-
¿Por qué el Estado no se dedica personalmente a la producción de medicamentos y así evitaría tener que tomar préstamos?
-
Mentiras en el costo del programa
-
según los integrantes de la Cicop, el Estado tendría en sus manos la posibilidad de abaratar los costos y de fomentar la producción nacional dado que “cuenta con la infraestructura y el personal para hacerlo”.
-
“La política actual del Gobiernos en materia de medicamentos tiene dos ejes, una es la de lo genéricos y el otro es el programa REMEDIAR, que en conjunto no dan respuesta a los requerimientos de remedios de la población o lo hace muy parcialmente a un costo social demasiado alto para el país, porque genera endeudamiento externo y porque el crédito del BID implica condicionamientos, como el de los lugares donde debemos comprar y la prohibición de poner en marcha los laboratorios estatales”
-
“Tenemos alrededor de 40 centros de producción de medicamentos que están trabajando por debajo de su capacidad real. Si en lugar de endeudarse el Estado invirtiera el dinero para reactivar la producción de genéricos en estas plantas, evitaría la fuga de cerebros y ahorraría muchísimo dinero”.
-
Bienes:
-
Obras: US$ 3.350.050
-
Consultoria:
-
Se facilitarán también recursos para consolidar la provisión de material y medicamentos esenciales y para la capacitación de profesionales de salud en el uso racional de medicamentos bajo el programa Remediar, conocido por sus altos niveles de eficiencia y transparencia para garantizar el acceso a medicamentos en el nivel primario de atención dentro del sistema de salud pública. Se ofrecerá capacitación a los trabajadores de los servicios de salud dentro del Programa de Médicos Comunitarios para reorientar sus conocimientos hacia la atención de la salud primaria.
-
El Ministerio de Salud de Argentina estará a cargo del proyecto.
-
El préstamo tendrá un plazo de 25 años, con un período de gracia de cinco años y medio y una tasa de interés ajustable. Los fondos de contrapartida local sumarán US$57.5 millones.
-
Rescatar a los laboratorios, que venían perdiendo ventas.
-
Rescatar al ministro, para garantizar su continuidad en la administración Duhalde-Kirchner.
-
Rescatar una fuente de negocios por izquierda”.
-
reconocer patentes para invenciones en todos los campos de la tecnología, con excepciones limitadas;
-
no hacer discriminaciones en lo que se refiere a la disponibilidad o disfrute de los derechos de patente;
-
otorgar los derechos de patente por un plazo no inferior a veinte años desde la fecha de la solicitud;
-
limitar el alcance de las excepciones a los derechos de patente y conceder licencias obligatorias sólo bajo determinadas condiciones;
-
hacer cumplir los derechos de patente de manera efectiva.
-
ALTERINI y LOPEZ CABANA, "Responsabilidad profesional: el experto frente al profano"
-
BUERES, Alberto, "Responsabilidad civil de las clínicas",
-
CANTAFIO, Fabio Fidel, Régimen de comercialización de medicamentos Publicado en Sup. Act 26/10/2006, 1
-
Conclusiones I Jornadas sobre Responsabilidad por Productos Farmacéuticos y Medicinales; Morón, 1986
-
CORREA, Carlos M., "Protección de la información confidencial y competencia desleal"
-
DONATO, Nora - CANTAFIO, Fabio F., "Elementos para el debate en torno a los medicamentos genéricos", LA LEY, 2003-A, 1222
-
Enciclopedia Jurídica Omeba, Buenos Aires, "Medicina"
-
Carlos Correa, Universidad de Buenos Aires - Integrando la salud pública en la legislación sobre patentes de los países en desarrollo - South Centre - Buenos Aires, Argentina - 2001
-
Informe especial - Medicamentos: publicidad y réditos económicos - HOY La Universidad, Periódico Digital (Universidad Nacional de Córdoba) - 1 de julio de 2004 - http://www.hoylauniversidad.unc.edu.ar
-
Argentina: “La aplicación y el respeto de la ley de patentes medicinales constituyen los requisitos para invertir”, según John Lechleiter, presidente y chief opperating officer (COO) de Elly Lilly - La Nación - Argentina - 5 de febrero de 2006
-
“La pelea por la ley de patentes” - Revista Médicos - Número 11 - Página 26 - http://www.revistamedicos.com.ar/numero11/pagina26.htm
-
Comentarios a la nueva ley de genéricos en medicamentos - Dr. Carlos Damián Renna - Asociación Médica Argentina - http://www.ama-med.org.ar
-
“La Cicop reclamó la producción pública de medicamentos” - www.inforegion.com.ar
-
“Se perdieron inversiones por $ 40 mil millones desde 1998” - Diario Clarín - www.clarin.com/diario/2002/06/21/e.01001.htm
-
“PBI 2003: La economía crecería el 7,2%” - www.dolarsi.com/informes/PDF/145.pdf
-
“La nueva crisis argentina” - María Laura García - Trabajo presentado para la cátedra de Macroeconomía, Facultad de Ciencias Económicas, Universidad Nacional de Córdoba, Argentina, Marzo de 2002 - www.econlink.com.ar/economia/tango
-
“Política actual de medicamentos en nuestro país: Un análisis del Programa Remediar” - www.fmed.uba.ar/depto/ddhh/remediar.doc
-
www.anmat.gov.ar
-
www.msal.gov.ar
-
www.remediar.gov.ar
-
www.lexisnexis.com.ar
-
www.infoleg.gov.ar
-
www.federicotobar.com.ar
-
www.sssalud.gov.ar
-
www.presidencia.gov.ar
-
www.bvs.org.ar
-
www.iadb.org
-
www.cancerteam.com.ar/poli145.html
Aunque la implementación de tales medidas han sido llevadas a cabo con el objetivo primordial de minimizar la grave situación social que atravesaba el país en ese momento, la política de medicamentos que, en términos generales, ha llevado el gobierno nacional en los dos últimos años, significa un avance fundamental en términos de mejorar el bienestar de la población en el largo plazo.
Prescripción de medicamentos por nombre genérico
Frente a la necesidad de acercar la oferta de medicamentos a la demanda de la población en un contexto de fuerte caída de los ingresos reales y el aumento del desempleo, se ha implementado, entre otras medidas, la prescripción de los medicamentos por su nombre genérico. Tal medida de política llevada a cabo, en un principio, a partir de la Resolución Ministerial 326/02, y luego con la sanción de la Ley 25.649, en agosto de 2002, reglamentada mediante el Decreto 987 de abril de 2003, establece que toda receta y/o prescripción médica u odontológica debe efectuarse expresando el nombre genérico del medicamento, seguida de forma farmacéutica, cantidad de unidades por envase y concentración.
Asimismo, el profesional farmacéutico deberá informar al paciente/consumidor sobre todas las marcas comerciales que contengan el mismo principio activo, en igual concentración, idéntica forma farmacéutica y la misma cantidad de unidades indicando en cada caso los distintos precios de estos específicos. De este modo, el paciente/consumidor podrá elegir la marca y precio del medicamento prescripto por el médico, no autorizando la sustitución de la droga prescripta por el médico.
Teniendo en cuenta que la utilización de los medicamentos constituyen una necesidad que en general no puede ser postergada, el gasto de un individuo en estos productos de características tan particulares, se convierte en un gasto fijo regresivo, teniendo una incidencia proporcionalmente mayor, a medida que cae el nivel de ingresos. La política de prescripción de medicamentos por nombre genérico, contribuye a reducir el precio de los medicamentos, debido a que se introduce un mecanismo de competencia entre las empresas productoras, por lo tanto la incidencia del gasto en esos bienes sobre los gastos totales de las familias, se ve reducida, permitiendo una significativa mejora en el acceso a los medicamentos.
La posibilidad de la implementación de la política de prescripción por nombre genérico y el reemplazo de los medicamentos en función de la elección por parte del paciente/consumidor, exige como requisito primordial garantizar la calidad de las distintas especialidades medicinales comercializadas en el mercado farmacéutico, en lo que hace a la seguridad y eficacia.
Con esta premisa el Ministerio de Salud de la Nación a través de la Administración Nacional de Medicamentos Alimentos y Tecnología Medica (ANMAT), asegura la calidad de los medicamentos sobre la base de lineamientos esenciales que aseguran la calidad de los fármacos.
Las Buenas Prácticas de Manufactura y Control es uno de los pilares principales en la construcción de la calidad de un producto, la verificación del cumplimiento de las referidas, tanto en establecimientos elaboradores como en establecimientos importadores, la liberación del primer lote y las inspecciones periódicas a los establecimientos antes mencionados, permite controlar la calidad y su homogeneidad, en los distintos lotes de los fármacos en esta primera etapa. Actualmente se analiza la implementación, en el año en curso de las GMP 2003 recomendadas por la OMS y anexos complementarios establecidos en el Sistema PIC-S. La toma periódica de muestras en el mercado en toda la cadena de comercialización y en los distintos puntos del país, es otro eslabón muy tenido en cuenta por la administración. A tal fin se cuenta con un programa de muestreo del mercado con un promedio de 3000 productos analizados/año.
El Sistema Nacional de Fármacovigilancia, permite la detección temprana de efectos adversos o inesperados en la etapa de uso extendido, tal situación genera la posibilidad de emitir nuevas recomendaciones, advertencias, restricciones de utilización y en algunos casos retirar del mercado aquellas especialidades medicinales que han demostrado tener una ecuación riesgo/beneficio desfavorable.
Asimismo, se dio especial relevancia al Programa de Pesquisa de Medicamentos Ilegítimos, a fin de evitar la comercialización de productos ilegales (falsificaciones, productos no registrados, productos mal llamados medicamentos magistrales ya que son producidos a escala industrial, provenientes del contrabando, vencidos, etc.). Este programa tiene un promedio de visitas de 2800 por año a establecimientos de los distintos puntos de comercialización. En los últimos 4 años se ha inspeccionados establecimientos radicados en todo el país.
La nueva Edición de la Farmacopea Argentina, es otras de las acciones tendientes a trabajar en la calidad de los medicamentos, ya que establece criterios y especificaciones de calidad que deben cumplir los medicamentos en temas referidos a pureza de materias primas, procesos de producción y controles entre otros que refuerzan las acciones mencionadas.
La política de prescripción de medicamentos por su nombre genérico y la consecuente posibilidad de reemplazo, exigió además de garantizar la calidad de los productos aprobados, analizar en profundidad las características particulares de los mismos, por lo que se entendió que para aquellas especialidades medicinales que contengan principios activos de estrecho rango terapéutico y por ende constituir el grupo de fármacos considerados de alto riesgo sanitario era necesario exigir otro estándar de calidad. Es así que en estos casos particulares, se desaconseja el reemplazo cuando no hayan demostrado, a través de ensayos aprobados por la ANMAT, su bioequivalencia con el producto de referencia.
Estas acciones emprendidas por el Ministerio de Salud de la Nación para facilitar el acceso a los medicamentos además, de destacar la actividad del profesional médico en la responsabilidad de constituirse en una herramienta fundamental para llegar a lograr el fin perseguido asesorando al paciente sobre las distintas alternativas terapéuticas y asimismo, jerarquiza el papel del farmacéutico, ya que es un profesional especializado en medicamentos que cuenta con la capacidad y autoridad para proponer el reemplazo de una determinada marca comercial por otra que contenga el mismo principio activo, asesorando y orientando al paciente/consumidor en su elección.
|
| Antes | Después | Var. % |
| Drogas Genéricas | 215 | 377 | 75 % |
| Presentación Comercial | 2645 | 3902 | 48 % |
| Medicamentos al 40% | 192 | 178 | - 7 % |
| Medicamentos al 70% | 3 | 112 | 3633 % |
| Medicamentos al 100% | 20 | 87 | 335 % |
Fuente: Unidad de Investigación Estratégica en Salud. Ministerio de Salud y Ambiente de la Nación en base a Superintendencia de Servicios de Salud.
Respecto a los medios de comunicación el Diario Clarín en noviembre del año 2003 posterior a la sanción y a los efectos de establecer el impacto económico respecto del empleo de ésta, de lo que deduce que la ley de medicamentos genéricos aumentó el empleo entre los farmacéuticos así como también se produjo una reactivación de la demanda en laboratorios nacionales. Lucio Di Matteo afirma que la ley de medicamentos genéricos cambió el mapa laboral de la industria farmacéutica. Mientras que los laboratorios extranjeros redujeron personal o lo conservaron con mucho esfuerzo y los nacionales con marcas fuertes mantuvieron sus dotaciones, los productores de genéricos las incrementaron sensiblemente.
La facturación de los laboratorios de genéricos treparía a casi 279 millones de pesos este año, mientras que en 2002 fue de 160,7. Este crecimiento se tradujo en duplicación del empleo: de casi 900 a más de 1.800 empleados, entre mayo de 2002 y la actualidad, según datos de la Cámara Argentina de Productores de Medicamentos Genéricos y de Uso Hospitalario (Capgen).
"La mayor parte de los operarios que tomamos habían sido despedidos por laboratorios multinacionales. También ingresaron profesionales en las áreas comercial y administrativa", señala José Luis Tombazzi, presidente de Capgen.
La "Ley de Promoción de la utilización de medicamentos por su nombre genérico" también produjo un efecto diferenciado entre profesiones. Los más perjudicados fueron los Agentes de Propaganda Médica (APM) o "visitadores médicos".
Mientras que los farmacéuticos prácticamente gozan de pleno empleo en Capital Federal y Gran Buenos Aires, y aumentaron su nivel de ocupación el 8% en un año.
"La ley establece que sólo el farmacéutico puede reemplazar una marca por otra de la misma droga; lo que provocó que quienes sólo trabajaban algunas horas en la farmacia ahora lo hagan todo el día. También hubo incorporaciones, porque no pueden cumplir turnos superiores a 8 horas y generalmente las farmacias están abiertas más tiempo", explica Marcelo Peretta, vicepresidente de la Confederación Farmacéutica Argentina (COFA).
"Los laboratorios nacionales con marcas reaccionaron con mayor flexibilidad de estrategias y precios que los importados, lo que les permitió mantener su facturación o incrementarla", dice Clara Suárez, directora ejecutiva de Cooperala, una de las cámaras que agrupa firmas de origen argentino.
Reacciones
Un ejemplo de esta tendencia es Raffo, que a pesar de tener marcas reconocidas y 105 años de vida, lanzó Tecnomax —el sildenafil (la droga del Viagra) con el precio más bajo del mercado— casi como un genérico, y en cuatro meses trepó del décimo al sexto puesto.
"Somos un laboratorio 'marquista' de toda la vida; esta fue una estrategia entre otras posibles, como mayor agresividad comercial, flexibilidad en los precios y lanzamiento de nuevos productos. La ley de genéricos nos favoreció en algunos aspectos y nos perjudicó en otros, por lo que apostamos a un mix de medidas", asegura Carlos Eraldo, gerente comercial de Raffo.
Entre agosto de 2002 e igual mes de 2003 las unidades vendidas por Raffo crecieron 23%; y la dotación de personal pasó de 460 a 540 personas (más del 17%).
"En los próximos meses incorporaremos alrededor de 20 personas para el área comercial, y 50 más en otros sectores", apuesta Eraldo.
Esto también se debe a que en su planta de Pocitos, San Juan, triplicaron su producción desde principios de año hasta ahora.
Otros laboratorios nacionales que crecieron son Baliarda, Temis Lostaló, Andrómaco y Elea. Por ende, según lo publicado, podemos advertir un fuerte incremento del mercado interno
El Diario El País publicó un aviso 3 años después, donde evaluó los diversos efectos colaterales de la sanción de la ley 25649 y todo el sistema de soporte para acompañarla. Afirma que éste configuró un ahorro importante para la población. No obstante un especialista encendió el alerta: “Si no son de calidad, son un peligro”.
El ahorro le ganó finalmente a la marca. Ya el 70% de los medicamentos recetados en la Argentina son pedidos por su nombre genérico, de acuerdo a un informe oficial difundido un día anterior a la publicación, a poco de cumplirse el tercer año de vigencia de la norma que instauró el sistema.
La proporción del alcance de esa modalidad se eleva al 79,1% en la zona de Buenos Aires y de la Capital Federal, uno de los porcentajes “más altos del mundo”, según se estableció en el estudio elaborado por el Ministerio de Salud.
El trabajo también reveló que en el primer semestre del año, casi el 90% de los medicamentos bajaron de precio “en términos reales”, debido al “nivel de aplicación de la prescripción por nombre genérico en todo el país”, indicó el trabajo.
De todas formas, afirman, hay que tener cuidado. Si bien la prescripción de medicamentos genéricos es una herramienta válida a la hora de comprar precios, debe ser hecha “con productos que sean de calidad y que brinden la seguridad de que son útiles para el paciente”, advirtió un especialista en farmacología y desarrollo de nuevas drogas.
El británico Atholl Johnston, quien visitó Buenos Aires para ofrecer una charla en la Sociedad Argentina de Hematología, afirmó que los genéricos “deben ser productos realmente de calidad” y deben tener “todos los avales detrás para darle seguridad a los médicos de que ese medicamento es útil y es bueno”.
“Cualquier pequeño y hasta insignificante cambio en la formulación de un medicamento, puede provocar grandes diferencias en la cantidad de droga que es enviada al sistema circulatorio”, advirtió.
Además, el especialista, quien se desempeña en la Barts and The London Quenn Marys's School of Medicine, remarcó que “hay una gran diferencia entre la prescripción por genéricos y la sustitución de un medicamento por un genérico”, y aseguró que esto último puede acarrear riesgos para la salud.
Johnston destacó que con ellos “lo que se persigue es contener los costos en la prescripción de los productos. Y reducir esos costos muchas veces es una necesidad”.
“Es una meta muy deseable de alcanzar, pero la única condición que se solicita es que los productos genéricos que se van a prescribir sean seguros, eficaces y tengan una consistencia en la producción, es decir que sea igual un lote con respecto a otros”, indicó.
El ministro de Salud, Ginés González García, advirtió que “existen leyes parecidas a la nuestra en varios países, pero en ningún lugar se observa el mismo nivel de aplicación. La ley española tiene un cumplimiento del 25%, y en Estados Unidos la prescripción no llega al 50%”.
Datos
• De acuerdo al trabajo oficial, la política de genéricos coincidió con la recuperación del consumo de medicamentos, que de un piso en 2002 de 277 millones de unidades, pasó a 439 millones que se proyecta para este año.
• Según datos de la última medición realizada por la Comisión Nacional de Programas de Investigación Sanitaria (Conapris), el 71% de las recetas que se elaboran en todo el país se realizan bajo el nombre genérico del medicamento.
• Además, se indicó que “en los últimos 5 meses y con una inflación estimada en 8% anual, los medicamentos sólo se incrementaron en un promedio del 3,7%, lo que implica una reducción promedio de precios en términos reales”.
• El 28 de agosto del 2002 fue sancionado el proyecto de ley de prescripción de medicamentos por su nombre genérico, que finalmente se convirtió en la ley vigente el 19 de setiembre.
• La norma fue incluida como parte de la Política Nacional de Medicamentos, que se completó con el Programa Remediar y el aumento de la cobertura de la Seguridad Social del 40 al 70% para pacientes crónicos, la desgravación de insumos críticos, el reordenamiento de la producción estatal, entre otras.
• Durante el primer semestre de 2005, del total de presentaciones que se comercializan en la Argentina (19.750), el 89,7% disminuyeron sus precios en términos reales, ya que “o mantuvieron sus precios nominales o experimentaron una reducción”.
• Finalmente, el estudio incluyó una encuesta elaborada por Análisis e Inteligencia de Mercado (AIM), que reveló que “el 92,5% de los entrevistados dicen estar de acuerdo con la nueva modalidad y sólo el 2,5% se muestra en desacuerdo”.
Entre los profesionales también se suscitaron diversos fueros de discusión sobre el impacto económico de la mencionada norma. Una de ellas se vio enmarcada en un encuentro organizado por la Asociación de Economía de la Salud (AES). “Impacto económico de la difusión de los medicamentos genéricos" fue el tema convocante del encuentro del 13 de agosto en la Fundación Isalud, en el que participaron como disertantes Joan Rovira Forns, Senior Health Economist del Banco Mundial, el Lic. Federico Tobar, Coordinador del programa Remediar y el Cdor. Carlos Vassallo, Vicepresidente de la AES. Rovira reseñó el rol del Banco Mundial en el sector farmacéutico internacional y describió el proyecto de creación de un Global Store, entidad multinacional que tendría como función la gestión de la compra de medicamentos por parte de grupos de países en vías de desarrollo. El Lic. Tobar analizó la conveniencia o no de la producción pública y destacó que en el escenario económico de los próximos años "para recuperar el acceso a los medicamentos el protagonismo del Estado es fundamental y la atención pública va a ser indelegable". Por su parte, el Cdor. Vassallo calificó la actual política de medicamentos como una "verdadera revolución", aunque admitió que "se necesitan instrumentos adicionales que el sistema de Salud no tiene".
Según informó el economista Joan Rovira, el Banco Mundial está cambiando su política con respecto a los medicamentos. Tomó una clara postura con respecto a los acuerdos Trips e incluso asesora a los países en desarrollo respecto al abordaje de este tema para su "mejor aprovechamiento", también está trabajando en distintos proyectos sobre "precios equitativos" entre los cuales se destaca la creación de un Global Store que gestione la compra de fármacos para grupos de países. Para conseguir precios equitativos existen dos posibilidades: Una, es conseguir precios ajustados a las posibilidades de cada uno de los países. Otra vía sería que se pagaran derechos de patente más bajos para que los fármacos se puedan producir en cada país con menor costo". En este marco se inserta el proyecto de creación de una entidad, propiedad de varios países, para negociar con las empresas. "Evidentemente, si este comprador representa a varios países, tiene mayor capacidad de negociación que si representa a uno solo, pero aparte de la negociación puede hacer como Brasil: plantear que en caso de no conseguir buen precio, producirá él mismo los medicamentos. El esquema que pensamos es pagar a la empresa innovadora un 10% del costo del fármaco en términos de royalties, pero que empresas de cualquier país lo puedan producir (hagan investigación o sólo las últimas fases del proceso) y que el Global Store le compre a cualquier empresa con sistema de concurso, entonces se genera la competencia. Al innovador se le paga para mantener el incentivo, pero no es el proveedor exclusivo. Este comprador haría también la distribución a los distintos países. Esto permitiría que o bien con sus propios fondos, o bien con fondos adicionales de donantes, como el Fondo Global para Sida y Malaria, los países puedan tener precios que permitan una mayor accesibilidad. Esto agrega además otro factor importante: la solidaridad. Este mecanismo exige segmentar el mercado, que los productos que se obtengan en un país en vías de desarrollo a menor precio no vayan a los mercados ricos, porque esto destruiría el sistema".
Finalmente, Rovira planteó la necesidad de pensar en esquemas regionales, ya que no muchos países pueden producir las distintas etapas de un fármaco, y sobre todo, no todos los países van a poder producir los principios activos. Incluso en el caso de que tuvieran licencias obligatorias, muchos no tienen capacidad técnica. En ese sentido propuso un acuerdo que tuviera en cuenta el proceso de producción.
Encrucijada
El Lic. Federico Tobar, Coordinador del Programa de Reforma de la Atención Primaria de la Salud (PROAPS) y responsable del Programa Remediar señaló que "la mayor preocupación del grupo que trabaja en políticas de medicamentos en el Ministerio de Salud de la Nación es la sustentabilidad de las acciones y la problemática del acceso".
El funcionario contextualizó la situación del mercado de medicamentos. "En todo el mundo el costo de la Salud ha aumentado en los últimos años. En particular hemos visto que creció la participación del gasto en medicamentos y más que el gasto en Salud en general. Otra particularidad terrible es que tiende a aumentar la regresividad de la financiación del gasto en medicamentos. Y tal vez lo más novedoso es que de las 1.393 patentes de medicamentos que se produjeron entre 1975 y 1993 es que, aunque hay innovación científica, no tiene repercusión sobre la Salud. De tres innovaciones sobre medicina tropical, que hoy es responsable del 70% de la carga de enfermedad en el mundo, cuatro tuvieron objetivos veterinarios. Tres de esos medicamentos fueron creados con objetivos bélicos. Solamente dos en realidad representan innovaciones realmente logradas con objetivos de salud pública. Que el laboratorio busque innovar donde haya rentabilidad hace a las reglas de juego de la economía, es el Estado el que tiene que incentivar para que haya generación de conocimiento que tenga aplicaciones concretas sobre la carga de enfermedad. Y el Estado ha fallado en esto y no digo sólo en la Argentina".
Impacto de la política de medicamentos
El Lic. Tobar presentó los resultados de una investigación multicéntrica desarrollada desde la CONAPI, Consejo Nacional de Políticas de Investigación, a finales de febrero de este año, en la que se encuestó a 8.000 pacientes, 2.000 médicos y a 900 farmacéuticos con control de recetas. Según reflejó el trabajo, el 55% incluyen el nombre genérico. "Es verdad que de ellas, más del 80% incluye el nombre comercial además del genérico -admitió-. Pero se consigue que de alguna forma se quiebre el monopolio".
"Del 45% restante -continuó el funcionario-, en el 27% de los casos, aun cuando se emitió solamente con el nombre comercial, el farmacéutico ofrece al paciente o el paciente le reclama al farmacéutico una alternativa del mismo medicamento genérico, con lo cual el volumen de utilización es mucho mayor que simplemente la cantidad de recetas que se emiten por nombre genérico. Pero además de eso hay un 13% de los casos en los cuales aunque el farmacéutico le ofrezca una marca alternativa al paciente, el paciente decide optar por la que el médico le indicó. Es decir, existió la posibilidad de que la decisión pase al consumidor, que es el objetivo de la política. Solo en un 5% la única decisión es la del prescriptor".
Aunque el disertante sostuvo que "ésta es una política absolutamente exitosa", se preguntó en qué medida es sostenible y en qué medida va a generar mayor acceso a los medicamentos. "Aunque los números iniciales son muy alentadores para nosotros, también tenemos que tener una lectura crítica que nos haga pensar que si no encontramos una solución global más sustentable del tipo de una agencia internacional o una alianza internacional, un mecanismo eficiente de adquisición, va a ser difícil lograr recuperar el acceso".
El Ministro de Economía, Roberto Lavagna señala que aun en un escenario de crecimiento sostenido del Producto Bruto del orden de un 3 a un 4% anual, en el año 2010 el desempleo se mantendría estructural y podría llegar a estar entre el 10 y el 15%. "Eso significa que la capacidad de recuperar el acceso vía farmacia a través de la Seguridad Social va a ser ínfima -reflexionó Tobar-. Es decir, tenemos que encontrar algún otro mecanismo porque puede ser que haya una recuperación de los ingresos de la Seguridad Social, que la Seguridad Social comience a sanear sus finanzas, pero es muy difícil que expanda su cobertura poblacional".
Estado productor vs. Estado comprador
Remediar es el otro mecanismo que sostiene la Política Nacional de Medicamentos. El funcionario planteó en este caso otra de las encrucijadas en las que se encuentra el gobierno: "¿El programa tendría que ser de producción pública o no? Y ¿en qué medida tiene sentido que sigamos tomando créditos internacionales y endeudándonos para garantizar el acceso a los medicamentos?". Es una problemática grave -señaló-. Entonces, en función de pensar escenarios alternativos de crecimiento económico y expansión, la primera conclusión a la cual llegamos es que para recuperar el acceso el protagonismo del Estado es fundamental y la atención pública va a ser indelegable".
Tobar planteó la hipótesis que estudia el Ministerio en el marco de la proyección económica que plantea el Dr. Lavagna: si existe un crecimiento de los registros de patentes, es decir, si llegamos al 2010 con un volumen significativo de medicamentos innovadores patentados, probablemente se puedan lograr dos mercados claramente diferenciados, uno de genéricos y uno de medicamentos innovadores. Si no existe, en el escenario más optimista puede haber algún incentivo para mantener la oferta en la medida que se mantenga una cuota de distribución pública, sino lo más probable es que vaya a haber un conjunto de productos que se van a discontinuar. Esto implica que se deberá mantener algún esquema de provisión pública". En este punto, según refirió el Dr. Tobar, el gobierno se planteó por qué comprarles a los laboratorios privados en lugar de incentivar la producción pública y tomar el ejemplo de Brasil, pero la respuesta que encontró este interrogante es que "en principio la política nacional de medicamentos tiene un compromiso prioritario con el acceso pero también con la calidad, entonces relajar los controles de la ANMAT o comprar desde el Ministerio medicamentos que no tienen el registro de un organismo que dependa del mismo Ministerio sería desandar un camino en el que se avanzó bastante en los últimos diez años. Me parece que la vía sería, en todo caso, si queremos incrementar el protagonismo de la producción pública, lograr una inversión en la capacidad de producción de los laboratorios públicos como para que todos alcancen los estándares de calidad de los privados". Sin embargo, el funcionario afirmó: "No sé si eso sería eficiente en términos económicos; cuando hay capacidad instalada, know how y trayectoria de producción. Me parece que lo más interesante sería discutir una división de trabajo coherente, de manera que los 40 laboratorios públicos que hay en las provincias, en los municipios, en las universidades, en las Fuerzas Armadas, produzcan un mix de productos que no sean exactamente lo mismo que el mercado puede producir igual o mejor, probablemente mejor".
Con este concepto concordó el Cdor. Carlos Vassallo, Vicepresidente II de la Asociación de Economía de la Salud, quien opinó: "Resulta bastante audaz pensar que los laboratorios públicos pueden llegar a producir medicamentos como alguna vez se planteó que era el gran negocio, que hasta los municipios pensaban poner un laboratorio en un garage. Hay que tener las prevenciones suficientes como para poder seguir asegurando los niveles de calidad. Estamos en un punto en que se hace necesaria una ley para establecer un control de calidad de los medicamentos disponibles, que controle a nivel nacional como provincial aspectos de producción, distribución y dispensa".
Por otra parte, señaló que aunque la Ley de prescripción de medicamentos por nombre genérico "pareció una verdadera revolución, se ha amortizado como política mucho más rápido de lo que uno podía llegar a pensar y hoy necesita de otros instrumentos adicionales que el sistema de Salud no tiene. En primer lugar, por su grado de fragmentación y por la escasa presencia del financiamiento público. Las políticas que se apliquen deberían considerar tres aspectos -concluyó el Cdor. Vassallo-: "Primero, no confundir gasto con precio y con costo de medicamentos. Segundo, no intervenir exclusivamente sobre uno de los sectores que integran la cadena sino trabajar con todos y tampoco hacerlo en forma cortoplacista pensando en los ciclos electorales sino tratando de pensar en el largo plazo y, finalmente, lograr optimizar la utilización de los medicamentos, que es uno de los desafíos que plantea la modernización de los niveles de calidad de nuestro sistema".
PROGRAMA REMEDIAR
Advertencia
Al adentrarse en el estudio del Proyecto Remediar el lector se va a encontrar con una postura positiva de amplio desarrollo. Es más, si se decide por ampliar sus conocimientos en la materia lo más frecuente es encontrar loas constantes al programa. No obstante ello cuando se encuentran críticas, éstas son de un tenor muy relevante. En alguno casos la vehemencia del autor es llamativa. Es decir, hay defensores acérrimos del plan y críticos de gran ímpetu.
Para tratar de analizar el tema de forma objetiva en este trabajo se optó por hacer un esfuerzo y tratar de equilibrar el debate de forma tal que quien lea pueda sacar sus propias conclusiones.
Metodológicamente, primero se procederá al desplegar la postura oficial y la de quienes consideran que el plan es una experiencia netamente positiva y ejemplar. En segundo lugar se procederá a exponer las posturas que no comulgan con tales aserciones. Como corolario emitiremos un breve dictamen acerca de la experiencia en curso.
La voz “oficial” y las versiones favorables
¿Qué es “Remediar”?
En la actualidad en el sitio web oficial del programa -www.remediar.gov.ar- se lo describe de la siguiente manera:
Remediar cumple con los objetivos de fortalecer el modelo de atención primaria y promover políticas saludables con gestión participativa y de asegurar el acceso a los medicamentos esenciales a la población más vulnerable. En especial para quienes están bajo línea de pobreza o no tienen cobertura de obra social.
Para alcanzar tales objetivos, Remediar se propuso proveer medicamentos esenciales buscando llegar a todos los Centros de Atención Primaria de la Salud del territorio nacional para su prescripción y suministro gratuito a la población objetivo.
En febrero de 2002 nace Remediar para hacer frente a la crisis sanitaria que atravesaba nuestro país y se establece como una herramienta fundamental de la Política Nacional de Medicamentos. Remediar se convierte en el instrumento con el cual el Ministerio de Salud de la Nación da una nueva dirección al PROAPS (Programa de Reforma de Atención Primaria de la Salud). En 2004 se establece el Plan Federal de Salud y el Programa se enmarca en él, respondiendo de esta manera a sus bases.
En este contexto y persiguiendo los objetivos señalados, en octubre del 2002 comienza la distribución gratuita de medicamentos esenciales, contando con una financiación hasta finales de 2004. Debido a la eficaz implementación del programa, la continuidad de Remediar está asegurada hasta fines de 2008 con el mismo presupuesto otorgado en sus inicios.
A partir de la experiencia que fue adquiriendo el Programa, de las diferentes evaluaciones y del análisis de la información que produce, empiezan a vislumbrarse problemáticas de salud a las que se les pueden dar respuesta en el marco del programa. Es así como comienzan a diagramarse diferentes estrategias que hoy cuentan con excelentes resultados.
Con referencia al funcionamiento de este complejo andamiaje el mismo sitio oficial nos ofrece la siguiente información:
El funcionamiento de Remediar es posible a través de diferentes líneas de acción que surgen como respuesta a las necesidades que se perciben cotidianamente en la constante relación entre los beneficiarios y efectores del programa.
En este sentido, y teniendo en cuenta la magnitud que ha ido cobrando Remediar en estos años, además de garantizar la entrega gratuita de medicamentos, se fueron implementando con éxito diversas estrategias desde las áreas que conforman Remediar, siempre persiguiendo los objetivos mencionados anteriormente.
Remediar, al incentivar la demanda directa en los CAPS, ha fortalecido la capacidad de los sistemas de salud provinciales y municipales para dar respuesta a los problemas de salud de sus habitantes jerarquizando la red asistencial.
Por otro lado, Remediar promovió e hizo efectiva la participación de la sociedad civil en diversas instancias del Programa promoviendo políticas saludables con gestión participativa para todos. Siguiendo este eje, se impulsa la capacitación de los equipos de salud en participación comunitaria, planificación local participativa y formulación de proyectos, brindando además financiamiento y asistencia técnica.
Al mismo tiempo ha colaborado en la mejora del sistema de salud a través de la complementación con la Política Nacional de Medicamentos en la promoción del acceso y el uso del nombre genérico y la promoción del uso racional de medicamentos, este último a través del Programa de Uso Racional de Medicamentos que cuenta con tres componentes que integran las diferentes instancias de trabajo: Capacitación al equipo de salud, Articulación institucional e Información a la comunidad.
Además, Remediar en el área de Formación de Recursos Humanos, continúa con la realización de Capacitaciones Operativas para todas aquellas personas que se desempeñan en los centros de salud de los diversos programas que se implementan desde Remediar.
Por último implementa el Programa Nacional de Desparasitación Masiva, que se está llevando a cabo en diversas regiones del país y que comprende dos grandes estrategias: la entrega del medicamento para tratar el problema de la parasitosis en más 1.200.000 de niños de 2 a 14 años de edad de todo el país y una efectiva campaña de educación sanitaria que contiene material de difusión con medidas preventivas para la población y la realización de acciones para promover hábitos saludables en torno a esta temática.
Más allá de las pomposas descripciones oficiales nos encontramos en condiciones de sostener que REMEDIAR es el mayor programa de provisión gratuita de medicamentos esenciales ambulatorios del mundo. El programa sigue una estrategia progresiva. Los medicamentos son distribuidos en botiquines a los Centros de Atención Primaria de la salud (CAPS). La composición y cantidad de los botiquines esta proyectada para que aumente paulatinamente, así como también la cantidad de CAPS que actuarán como efectores del programa.
Evolución histórica del programa
La implementación de Remediar se dividió en dos etapas: La primera, lanzada el 21 de octubre de 2002, ha cubierto 2.000 CAPS en 24 jurisdicciones, y la segunda lanzada el 25 de marzo de 2003, se extenderá hasta fines del año 2006, cubriendo un total de 5.389 CAPS.
Durante su primera etapa, Remediar trabajó con dos modelos de botiquines, como consecuencia de dificultades encontradas en el proceso de compra de los medicamentos:
En esta segunda etapa se distribuyen botiquines conteniendo 56 presentaciones, adecuados a los motivos de consulta propios de las regiones. El actual vademécum consta de 56 medicamentos de los cuales hace meses se compran 48, destinados a abarcar el 80% de las patologías prevalentes, según informes oficiales.
Actualmente el plan se encuentra prorrogado y con fondos disponibles hasta fines de 2008.
Los controles en la distribución
En materia de control, el programa fue diseñado de manera que se garantice la eficiencia y transparencia en el uso de los recursos. Tiene un riguroso control de gestión, por un lado, a través de información generada por el programa. Cada consulta médica da lugar a un registro en una planilla de recetas. En la misma figuran los datos del beneficiario, el diagnóstico, el medicamento y las cantidades prescriptas. Cuando el beneficiario reciba el medicamento deberá firmar su conformidad en la misma receta. Esa información será procesada por Remediar y permitirá monitorear el programa.
Los botiquines son distribuidos directamente a los Centros de Atención Primaria de la Salud. Esto permite establecer un sistema uniforme de entrega en todas las provincias y municipios de la Argentina, garantizando su seguimiento y control. Para ello se ha realizado una licitación nacional de operadores logísticos habilitados por el ANMAT para transportar medicamentos. La firma adjudicada se hace cargo del armado y distribución de los botiquines y dispone de un sistema de rastreo satelital y un sistema de códigos de barras permite el seguimiento permanente de cada botiquín.
Para determinar cuántos botiquines corresponden a cada provincia, en el ámbito del Consejo Federal de Salud -COFESA-, se estableció un índice que fija con criterios sanitarios objetivos la participación que corresponde a cada provincia sobre el total de botiquines.
Cada provincia seleccionó los centros de salud para la recepción de los botiquines considerando la cantidad de consultas mensuales que los mismos realizan. Los botiquines han sido diseñados para atender alrededor de 300 consultas médicas. Con el programa en pleno funcionamiento, Remediar repondrá mensualmente los botiquines en función de las consultas realizadas por cada centro.
Adicionalmente, cuenta con un equipo de auditores propios que recorren permanentemente los CAPS y supervisan los stocks disponibles en cada centro y el cumplimiento de los procedimientos. La auditoria se complementa con visitas a los hogares de los beneficiarios para verificar que los mismos hayan recibido la medicación en tiempo y forma.
En tercer lugar, Remediar cuenta con una red de control social directo a través de un convenio con Cáritas y Cruz Roja. A través de sus voluntarios estas entidades visitan periódicamente cada uno de los centros de salud e informan respecto a dificultades e irregularidades.
Además, se buscó promover la articulación con otros programas sociales y sanitarios y la participación social. Para esto último se estableció una Comisión Asesora Intersectorial integrada por organizaciones no gubernamentales con representación en todo el ámbito nacional, junto a las confederaciones médica y farmacéutica, así como representantes de los otros ministerios del área social de la Nación.
Impacto social del programa
La participación del gasto en medicamentos varió significativamente entre 2003 y 2005. Mientras que en el 2003 el peso del gasto en medicamentos sobre el total de gasto en salud de los cubiertos sólo por el sistema público era del 62%, para el 2005 esta participación habría descendido al 39%.
A su vez, en la compra de los medicamentos hubo un ahorro, con relación al precio de venta minorista del orden del 650%.
La población que consulta en los centros de salud tiene acceso gratuito e integral a los medicamentos incluidos en el botiquín Remediar. Para ello deberá consultar al profesional en el CAP correspondiente a su lugar de residencia. Si el médico le prescribe un medicamento el mismo le debe ser suministrado directamente en el CAP en cantidades acordes a las dosis y duración del tratamiento prescripto.
El Programa Remediar ha tenido un alto impacto redistributivo sobre la población de nuestro país. Sobre un total de 32,7 millones de recetas Remediar se identificaron 13,2 millones de beneficiarios distintos (personas únicas).
Existen unas 2,5 millones de recetas que no pudieron ser procesadas por no constar en ellas o bien el documento o bien el nombre de la persona.
Gráficamente:
A partir de la información de la encuesta realizada por el SIEMPRO puede estimarse la importancia de REMEDIAR en relación al ingreso de los beneficiarios:
• Valorada a precios de mercado, la dación hecha por REMEDIAR en promedio importa una transferencia equivalente al 24% del ingreso per cápita familiar de los beneficiarios.
• Llega a representar un 41% del ingreso, en el caso de los beneficiarios bajo la línea de indigencia (71% del total de beneficiarios).
El programa ha tenido una adecuada focalización de la población objetivo del mismo:
• 94% de los beneficiarios de REMEDIAR son personas bajo la línea de pobreza.
• 71% de los beneficiarios de REMEDIAR son personas bajo la línea de indigencia.
• El ingreso medio per cápita familiar del 6% de los beneficiarios que no son pobres, se encuentra apenas por encima de la línea de pobreza.
Implementación
A continuación se transcriben algunas preguntas publicadas en la web oficial bajo el título de “Preguntas frecuentes”. Esta trascripción responde a fines netamente metodológicos para entender de una forma más acabada el funcionamiento del programa.
¿Todos los centros y salas barriales dispensan los medicamentos del programa REMEDIAR?
Existen más de 6000 centros bajo Programa en todo el país. Puede consultarse el listado de centros y salas en los municipios; a través de la línea telefónica de REMEDIAR (0-800-666-3300).
¿Pueden recibir medicamentos todos los pacientes asistidos en el centro de salud o "salita" del barrio?
Sí, todos los que hagan una consulta médica en el centro o "salita" y, como resultado de ella, les serán recetados los medicamentos incluidos en el programa.
Yo tengo obra social, pero no puedo pagar los medicamentos. Si me atiendo en el centro de salud del barrio, ¿me dan los remedios?
Sí, le corresponde recibirlos. REMEDIAR es para todos. Ahora bien, se trata de un programa de emergencia y, como tal, está prioritariamente dirigido a los más necesitados. Por eso, apelamos a la solidaridad. Si usted está en condiciones de comprar el remedio, por favor, piense que otra persona puede no tener acceso. Pero si usted no puede comprarlos, le corresponde acceder a los medicamentos de REMEDIAR, aunque tenga obra social u otra cobertura.
¿Pueden pedirse medicamentos por cuenta propia, o cuando fueron recetados por un médico particular?
No. Los medicamentos de REMEDIAR sólo se dispensan como resultado de una consulta en el centro de salud pública, y siempre que ésta concluya con la receta del médico de la sala.
¿Es necesario que me revise el médico del centro de salud o sala del barrio para que me entreguen medicamentos de REMEDIAR?
Sí, es imprescindible. Remediar también tiene por objetivo el uso racional y prudente de los medicamentos, que se favorece mediante la consulta y el diagnóstico del médico.
¿Se exige la presentación del documento de identidad a los beneficiarios?
Se solicitará su presentación, pero no se exigirá como condición para la entrega de los medicamentos. En la receta que usará el médico deberán incluirse sus datos básicos, como nombre, edad, sexo, e incluso documento de identidad. Si usted no tiene documento, debe ser aclarado en la receta; pero ello no impedirá que se le entregue el medicamento.
¿Pueden los médicos prescribir medicamentos en cualquier receta?
No. Deben utilizar el formulario único de receta que es provisto por el Programa REMEDIAR. De lo contrario, los remedios no serán repuestos al centro de salud. La receta de REMEDIAR es indispensable para el control y la continuidad del programa.
Estoy recibiendo tratamiento por una enfermedad crónica. ¿Puedo conseguir los medicamentos a través de REMEDIAR?
El programa provee algunos medicamentos para enfermedades crónicas, como la diabetes, la hipertensión, el asma, la epilepsia, la gastritis, el hipotiroidismo y la artritis. Si usted tiene otra patología conviene que averigüe por la cobertura de su caso en otros programas específicos que pueda haber en su área.
¿Pueden entregarse medicamentos en lugares distintos a los centros y salas barriales?
En su gran mayoría, los medicamentos serán entregados a través de los centros y salas de salud en los barrios. Puede ser que algunos remedios sean suministrados por médicos radiantes (que visitan a los pacientes en zonas rurales o muy alejadas de los centros de salud), siempre después de la revisión del paciente y de la prescripción con receta de REMEDIAR.
Hay algunos hospitales que reciben medicamentos, ¿por qué?
Sólo se entregarán remedios en algunos hospitales pequeños, donde predomina la atención primaria, y siempre que no haya salitas barriales o centros periféricos que puedan hacerlo en su lugar.
¿Qué debe hacerse si se venden medicamentos de REMEDIAR, o se distribuyen en otros sitios que los autorizados, o se entregan sin cumplir con los requisitos exigidos por el programa?
Denunciarlo. Hay distintas vías para realizar denuncias: el consejo local del centro o salita de salud, el municipio, la sede de Caritas o de Cruz Roja más cercana. También puede comunicarse gratuitamente con la línea directa de REMEDIAR (0-800-666-300), o a través del correo electrónico a remediar@proaps.gov.ar
¿El paciente debe firmar la receta?
Sí, pero sólo en el momento de recibir los medicamentos.
¿El paciente debe regresar por más medicamentos para completar un tratamiento?
No. En la consulta médica deben entregarle los medicamentos para el tratamiento completo. No deben faltar ni sobrar medicamentos. Ahora bien, si los síntomas persisten, usted debe volver a consultar al médico, y si éste le receta otros remedios -o más de los mismos-, usted volverá a recibir la medicación en las cantidades adecuadas.
¿Cuándo hay que completar el Formulario de Receta R?
Cuando se prescribe y entrega un medicamento o insumo que figura en el listado del Formulario R, y que es provisto por el Ministerio de Salud de la Nación a través de programas nacionales, o bien por la provincia o el municipio. Es decir, si el centro de salud recibe medicamentos e insumos de la provincia o del municipio, los que formen parte del vademécum de Remediar sólo podrán ser entregados al beneficiario contra presentación del Formulario de Receta R correctamente completado.
¿Cuánto cuesta el Plan Remediar?
Según información que consta en el sitio web del BID el costo total del programa en sus inicios asciende a US$ 167 millones, que será financiado mediante un aporte local equivalente a US$ 77 millones y un préstamo del BID de US$ 90 millones, para los 18 meses subsiguientes .El aporte local esta a cargo íntegramente del gobierno nacional.
El crédito en cuestión fue aprobado el 11 de agosto de 1999 y tiene fecha de contrato el día 2 de marzo de 2000. A fecha 30 de septiembre de 2007 se había realizado un efectivo desembolso por US$ 71.076.274 quedando aún pendientes US$ 18.923.726; esto concluyó en que si bien el plan original se concibió para se financiado hasta fines del año 2007 “Debido a la eficaz implementación del programa, la continuidad de Remediar está asegurada hasta fines de 2008 con el mismo presupuesto otorgado en sus inicios”.
El crédito en cuestión está registrado en la institución bajo la denominación 1193/OC-AR. La fecha de cancelación total es el día 2 de marzo de 2020 siendo su costo total de US$ 123.097.498 (US$ 90.000.000 de capital y 33.097.498 de intereses). La primer cuota de pago tuvo lugar el 2 de septiembre de 2007 -fuente: IDB Debt Service -.
Postura crítica
Si bien estadísticamente el programa parece netamente exitoso, existen varias voces que se alzan en forma de crítica. Los principales puntos son los siguientes:
A continuación pasaremos a analizar con mayor profundidad cada punto:
1.-¿A quién le compra el Remediar?
Según un relevamiento realizado por la Cátedra Libre de Salud y Derechos Humanos, y profesionales vinculados al área de la salud, en Marzo y Abril de este año en los Centros de Atención Primaria: Confluencia (Neuquén), ciudad de Córdoba, Wilde, Gelpi, Palomar, y tomando como datos la participación en las ventas de cada uno de los medicamentos.
Al inicio del Plan Remediar, el origen de laboratorios que vendían sus medicamentos era de diversos países, hoy podemos decir que en su mayoría son empresas de origen nacional.
Esta mirada transversal es sólo un dato para ver cómo es la participación actual de los laboratorios privados que venden al Plan Remediar sus productos.
Laboratorios que venden al plan Remediar según origen:
La composición de laboratorios nacionales y extranjeros que abastecen al Plan Remediar en una muestra transversal, de los Centros de Atención Primaria arriba nombrados. No se habla del volumen de dinero facturado por cada uno.
A pesar de estar registrados como "de capital nacional", muchos de los laboratorios están integrados con capitales de grandes transnacionales. El laboratorio Sandoz pertenece al Grupo Novartis; G&M es de Parke Davis. Richmond ha integrado capitales holandeses para la construcción de una planta en Pilar.
La venta por parte de laboratorios que responden a capitales extranjeros produce un doble perjuicio; se usa dinero ajeno en gran parte (préstamo del BID) para comprar medicamentos producidos por laboratorios que llevan sus ganancias al extranjero.
2.-¿Por qué el Estado no se dedica personalmente a la producción de medicamentos y así evitaría tener que tomar préstamos?
“El plan pasó por alto lo más interesante, que hubiese aniquilado el negociado: la inversión de una pequeña fracción de ese dinero en expandir la producción pública de medicamentos de buena calidad, según normas internacionales, hubiese logrado dar más trabajo local, jerarquizar instituciones del estado, bajar los precios de la licitación en decenas de veces, y eliminado las comisiones por la "gestión" del préstamo”.
Fuente: “Plan Remediar”; Buenos Aires, febrero de 2005, escrito por el Dr. Pedro M. Politi
-Oncólogo clínico, Equipo Interdisciplinario de Oncología. Profesor Adjunto, Segunda Cátedra de Farmacología, Facultad de Medicina, UBA. Asesor de Salud, Bloque 19 y 20, Legislatura de la Ciudad de Buenos Aires-. En esta crítica el profesional argumenta que
A su vez, la Asociación Sindical de Profesionales de la Salud de la Provincia de Buenos Aires (Cicop) entabló un reclamo análogo que fue publicado en www.inforegion.com.ar el día Domingo de 20 de Noviembre de 2005.
La producción pública de medicamentos, consistiría en una medida que a su entender “ahorraría dinero y evitaría la fuga de cerebros” y que no se toma porque, según ellos, el ministro de Salud de la Nación, Ginés González García, “es un hombre del Banco Mundial”.
El secretario de Prensa del gremio y titular de la delegación de Lomas de Zamora, Pablo Torres, entre otras cosas le dijo a Info Región que:
3.- Mentiras en el costo del programa
Quizás este sea el punto de mayor controversia del programa (y del que más se ha escrito al respecto). Como se dijo anteriormente el proyecto de forma oficial estaba pensado en un costo de US$ 167 millones, de los cuales el BID facilitaría US$ 90 millones en forma de un préstamo y el gobierno nacional aportaría US$ 77 millones.
La Cátedra Libre de Salud y Derechos Humanos de la Facultad de Medicina de la Universidad de Buenos Aires en su trabajo de investigación titulado “Política actual de medicamentos en nuestro país: Un análisis del Programa Remediar” sostiene que el costo es muy superior y ha sido escondido bajo partidas con nombres ajenos al programa pero que se vinculan de forma indirecta.
“Podemos decir que el costo total hasta el día de hoy es de US$ 233,784 millones del cual la fracción que otorga el BID es de US$ 140.000.000 que se suma a la deuda externa, y la que invierte el Estado Argentino como contraparte es de US$ 93,784 millones”.
El préstamo OC-AR 1193/3 del BID en su hoja llamada “Plan de Adquisiciones” tiene previsto gastar para el año 2005:
Entre los puntos más destacados de este ítem tenemos:
. Medicamentos Esenciales para CAPS con US$ 30.000.000
. Medicamentos (crónicos y esenciales) US$ 7.049.728
. Insumos Medicinales US$ 337.500
. Operador Logístico: distribución de botiquines US$ 2.960.000 lo que corresponde a un total de US$ 40.347.228 en medicamentos y distribución.
El resto de este punto asciende a un total de US$ 6.652.500.
Y esta distribuido en:
. Equipamiento básico para CAPS US$ 1.000.000
En Córdoba:
. Estaciones de trabajo en USF y ctorios. Etapas 1 y 2. US$ 904.000
. Dispositivos Móviles (USF) US$ 1.016.000
. Amp. Base de datos, servidores etc. US$ 3.465.000
. Comunicación Social. US$ 150.000
. Equipamiento de gestión US$ 117.500
Todo hace un total de US$ 46.999.728
Este punto hace referencia a la adecuación de los CAPS en distintas localidades de la Provincia de Córdoba, ver anexo
También en dicha provincia debemos considerar:
. Campaña de difusión US$ 600.000
. Auditoria de calidad US$ 143.5000
. Reconversión de RRHH US$ 3.756.000
. Adecuación de infraestructura US$ 3.372.500
. Capacitación de Sistemas US$ 202.000
. Becas US$ 827.586
. Pasajes y viáticos US$ 95.172
Lo que corresponde a un total de US$ 12.346.808
Este punto se divide en dos ítems:
. Consultores Individuales: para medicamentos y para la reforma de APS Córdoba suman US$1.216.063
. Firmas Consultoras: en Córdoba, para difusión, auditoria de calidad, capacitación de sistemas, estudios de fortalecimiento institucional, diseño de campaña integral de comunicación, mecánicas de comunicación, etc. Esto comprende la suma de US$1.933.000
Que hacen un total de US$3.149.063
Que entre todos los conceptos hacen un total de US$ 62.495.599 (gastos para el año 2005)
De este documento se desprende que lo destinado del crédito a medicamentos y distribución es el 64.56% para este año. Veremos entonces que el porcentaje destinado a medicamentos fue disminuyendo año a año obteniendo un promedio de 69.7%. Ver tabla I
Ahora bien, si estimamos lo que dice el gobierno respecto de que está distribuyendo 14.000 botiquines mensuales a todos los CAPS, estos sumarian 168.000 botiquines al año. Entre el gasto de medicamentos y su distribución, cada botiquín tendría un costo de US$ 240,16.
También del acuerdo con el BID se desprende que el Estado Argentino, este año, erogará el 40% del total del Plan Remediar, en el ítem de medicamentos lo que asciende a una suma de 13.407.419,6 US .
Si este programa se desarrolla desde octubre de 2002, y según las autoridades finalizará en noviembre del año 2006, tendremos que el Estado como contrapartida al préstamo pagó 93.784.000 millones de dólares a laboratorios privados, para comprar medicamentos, distribuirlos, consultorías, etc. Y no invirtió siquiera el 10% de esa suma en recursos propios para equipar a los Laboratorios Públicos. Tampoco prevé el convenio con el BID inversiones importantes del Estado para que aumente la producción de los Laboratorios Públicos Nacionales.
Claramente se ve que el Plan Remediar funciona entonces como una transferencia de recursos del sector estatal a la industria privada actuando como salvataje de la misma en la crisis del mercado público y privado de medicamentos de nuestro país. La financiación fundamental se materializa como deuda externa cuyo mecanismo de pago es la socialización de este pago por toda la población a través del producto social transformado en recursos fiscales que el Estado usa para pagar a los acreedores internacionales (FMI, BID, Banco Mundial etc).
Con base en esta información resulta interesante observar que el día 17 octubre, 2007 se emite un comunicado del BID que se titula: “BID aprueba US$230 millones para fortalecer estrategia de atención salud primaria en Argentina”
Entre otras cosas se sostiene que :
Teniendo en cuenta que ya está en ejecución un préstamo que debería cancelarse por completo en 2020 y que se empezó a pagar en 2007, que en ese mismo año se apruebe otro préstamo de mayor importe y a largo plazo, suena a que las críticas del punto anterior no han sido escuchadas.
Si ese dinero no es utilizado responsablemente seguiremos asistiendo al fenómeno de emparchar el presente hipotecando el futuro.
Por otro lado el monto tan superior al del plan anterior despierta la duda acerca de si el informe de la Cátedra Libre de Salud y Derechos Humanos no estará en lo correcto.
Llegado a esta instancia cabe preguntarse si no le asistirá razón al Dr. Politi que en otro fragmento del ya citado ensayo titulado “Plan Remdiar” sostiene que: “El plan remediar rescata a los laboratorios, comprometiendo más deuda externa”.
Desarrollando la idea el oncólogo argumenta; “El plan Remediar consistió en la compra de medicamentos considerados "esenciales" por el ministerio, mediante licitación pública, pagado con préstamos de organismos multilaterales de crédito (BID), en un país en default, a cubierto de decretos presidenciales (Duhalde; emergencia económica; emergencia sanitaria) le permitió y permite al ministro contratar sin respetar las normas de procuración del estado, pagar por adelantado (de ahí viene! Comprometió fondos y pagó por adelantado!!!) y en caso de conflicto, dar prioridad a los reclamos del BID y de la agencia intermediaria (OPS, Oficina Panam. De la Salud, con sede en Washington, dirigida por una funcionaria argentina, propuesta por Ginés G. García)”.
En esta línea de ideas el médico llega a una cruda conclusión:
“El plan Remediar es un "plan rescate"!, como el de Tucumán:
PATENTES
La industria farmacéutica es uno de los mercados más poderosos de alcances internacionales. Alrededor de ella se entretejen intereses políticos y económicos. Se trata de una de las industrias económicamente más poderosa sobre el planeta Tierra, en la que tan sólo 25 empresas controlan cerca del 50 por ciento del mercado mundial de medicamentos.
El mercado de medicamentos es un mercado oligopólico, es decir, altamente concentrado, con poder para fijar precios y profundamente innovador. Según la teoría económica, el oligopolio es una estructura de mercado en la que sólo hay unos cuantos vendedores importantes que acaparan casi toda la producción de la industria. En el sector de medicamentos esto se traduce en que pocos laboratorios productores tienen mucho poder de mercado.
No obstante, se suman otros factores que brindan una impronta diferente al desarrollo de la industria farmacológica. La necesidad de innovación permanente y la protección de los derechos de propiedad y patentamiento implican, a su vez, fuertes inversiones en investigación para el desarrollo de nuevas drogas. Esto hace que el mercado de los medicamentos sea fuertemente competitivo y se encuentre embarcado en un proceso de crecimiento continuo.
En este punto es necesario detenerse y aclarar algunas cuestiones. En primer lugar, la competitividad exige diferenciarse, y para lograrlo la innovación debe ser permanente. La empresa gana rentas extraordinarias por estar a la vanguardia. Cuando alguien entra con una nueva droga al mercado fija el precio, siempre por encima de los ya existentes. Sin embargo, a medida que se generaliza a través de la copia -principal forma de progreso técnico para muchos países- el precio decrece. La diferencia que hay entre el precio inicial del innovador y el precio de largo plazo del medicamento es lo que configura la ganancia extraordinaria.
El mercado farmacológico funciona de la siguiente manera: un laboratorio saca un nuevo producto al mercado y ejerce el monopolio del mismo hasta la caducidad de la patente. Una vez que la producción de ese medicamento se generaliza disminuye el precio. Sin embargo, la empresa sigue gozando de un mayor flujo de ganancias que sus competidores gracias a la oportuna inversión en publicidad.
Otra forma de diferenciación la brindan la marca comercial o reputación del laboratorio, la publicidad, la presentación y el envase.
El sector farmacéutico es uno de los principales usuarios del sistema de patentes. Diversos estudios han demostrado que la industria farmacéutica "de investigación" asigna una particular importancia a la posibilidad de obtener patentes en escala mundial. La protección perseguida, como se verá, no sólo se refiere a nuevas moléculas sino a un amplio rango de desarrollos tales como formas en que se presentan los compuestos químicos, procesos, formulaciones farmacéuticas, formas de administración y, donde ello es admitido por la ley, usos de productos conocidos. Así, si bien el número de nuevas entidades químicas aprobadas por la Food and Drug Administration (FDA.) de Estados Unidos declinó a veintisiete en el año 2000, comparado con alrededor de sesenta en 1985, el número de patentes concedidas en la principal clase de patentes para nuevas composiciones farmacéuticas (424) fue de 6730 en 2000.
El creciente número y la complejidad técnica del patentamiento en el campo farmacéutico plantean particulares desafíos a las oficinas de patentes latinoamericanas, especialmente a aquellas de países que no reconocían hasta hace poco patentes de productos farmacéuticos, o de éstos y procesos de elaboración, como en el caso del Brasil. En este último país se ha dispuesto la intervención del Ministerio de Salud en el examen de las solicitudes que versan sobre invenciones farmacéuticas, a efectos de mejorar la calidad de aquél, teniendo en cuenta las repercusiones de las patentes sobre la salud pública.
El Acuerdo sobre los Aspectos de los Derechos de Propiedad Intelectual relacionados con el Comercio (ADPIC) exige que todos los Estados Miembros de la OMC adapten sus legislaciones a las normas mínimas que establece el propio Acuerdo, dentro de períodos de transición estipulados. Dar cumplimiento al Acuerdo reconociendo o reforzando la protección de los derechos de propiedad intelectual (DPI) de los productos y procedimientos farmacéuticos plantea un reto especial a los países en desarrollo. La manera en que se lleve a cabo la reforma legislativa necesaria puede tener un impacto significativo en las políticas de salud pública, y particularmente en el acceso de la población a los medicamentos.
Dentro de las limitaciones que imponen los compromisos internacionales, y señaladamente el Acuerdo sobre los ADPIC de la Organización Mundial del Comercio, la legislación sobre patentes de los países en desarrollo debería: a) estar pensada para servir los intereses de todos los grupos sociales, y b) tener en cuenta los objetivos de la política de salud, y en particular las necesidades de la población de menores recursos.
Debe recordarse que las patentes farmacéuticas han suscitado por largo tiempo un intenso debate. Por su propia naturaleza, las patentes conducen a precios superiores a los que regirían en ausencia de protección y afectan el acceso a los medicamentos, especialmente por parte de los pobres. A diferencia de lo que ocurre en los países industrializados, la mayor parte del gasto en medicamentos es realizado en América Latina por el propio paciente, y no por los sistemas de seguridad social. La Conferencia Ministerial de Doha de la OMC. aprobó la Declaración de Doha relativa al Acuerdo sobre los ADPIC. y la Salud Pública (noviembre de 2001). La discusión de esta declaración fue una de las cuestiones más sobresalientes de la conferencia y el primer resultado de un proceso que comenzó en 2001 cuando, a petición del Grupo Africano, el Consejo de los ADPIC. acordó tratar específicamente la relación existente entre el Acuerdo sobre los ADPIC. y la salud pública.
Dicha declaración confirmó el derecho de los países miembros de la OMC. a proteger la salud pública en el contexto del Acuerdo ADPIC. y a utilizar plenamente la "flexibilidad" que deja el Acuerdo en diversos aspectos, tales como el de admitir importaciones paralelas o conceder licencias obligatorias.
Declaración de Doha relativa al Acuerdo sobre los ADPIC. y la Salud Pública, párr. 4º:
"4. Convenimos en que el Acuerdo sobre los ADPIC. no impide ni deberá impedir que los miembros adopten medidas para proteger la salud pública. En consecuencia, al tiempo que reiteramos nuestro compromiso con el Acuerdo sobre los ADPIC., afirmamos que dicho Acuerdo puede y deberá ser interpretado y aplicado de una manera que apoye el derecho de los miembros de la OMC. de proteger la salud pública y, en particular, de promover el acceso a los medicamentos para todos”.
"A este respecto, reafirmamos el derecho de los miembros de la OMC. de utilizar, al máximo, las disposiciones del Acuerdo sobre los ADPIC., que prevén flexibilidad a este efecto".
Una de las principales flexibilidades que deja el Acuerdo sobre los ADPIC. es la determinación de los estándares con que pueden aplicarse los requisitos de patentabilidad que prevé el art. 27.1 del Acuerdo.
Existe un amplio consenso en cuanto al papel que las patentes y los DPI pueden desempeñar en el fomento de la investigación y el desarrollo (I+D) aplicados al campo de la salud, sobre todo en los países más adelantados. Las patentes se consideran particularmente importantes, dado que la I+D entraña costos y riesgos elevados, y al mismo tiempo puede desembocar en invenciones de utilidad potencial para todos los países. Se reconoce asimismo que el nivel de protección otorgado a las invenciones puede influir en la inversión extranjera, la transferencia de tecnología y la investigación (especialmente sobre programas conjuntos de investigación y la investigación orientada a atender necesidades locales). Las patentes, al conferir un monopolio autorizado por el gobierno durante un tiempo limitado, incentivan y recompensan las invenciones útiles.
Pero los DPI tienen costos de precio y de competencia. En el sector de la salud, donde la falta de acceso asequible al tratamiento o a los productos farmacéuticos puede tener consecuencias de vida o muerte, las condiciones que determinan el acceso a las medicinas, y entre ellas el precio, son cuestiones críticas, sobre todo para los segmentos de ingresos bajos de la población. Aun reconociendo que los DPI no son el único factor pertinente, parece claro que la manera en que se establezcan y apliquen los DPI puede tener un impacto significativo en el acceso a las medicinas. Por lo tanto, todo régimen de DPI debe establecer un equilibrio entre la creación de incentivos a la innovación y el interés de los consumidores en que los bienes protegidos estén disponibles y accesibles.
El Acuerdo sobre los ADPIC ha introducido un marco internacional nuevo e importante para los DPI, que a su vez encierra serias repercusiones para el sector de la salud. El Acuerdo especifica obligaciones detalladas en lo tocante a la protección de las invenciones, entre ellas:
El Acuerdo sobre los ADPIC no establece, sin embargo, una regulación internacional uniforme, ni siquiera requisitos legales uniformes. Los Estados Miembros de la OMC están obligados a respetar las normas mínimas del Acuerdo, pero también disponen de un margen de maniobra considerable para configurar su propia normativa sobre patentes y otros aspectos de la propiedad intelectual de acuerdo con las características de sus ordenamientos jurídicos y sus necesidades de desarrollo. A la hora de poner en práctica las disposiciones del Acuerdo, los Estados Miembros de la OMC pueden legítimamente adoptar reglamentos que aseguren un equilibrio entre las normas mínimas de protección de los DPI y el bien público. Además de eso, pueden tomar medidas que favorezcan el bienestar social y económico (artículo 7 del Acuerdo sobre los ADPIC), por ejemplo las que sean necesarias para proteger la salud pública, la nutrición y el interés público en sectores de importancia vital para su desarrollo socioeconómico y tecnológico. Igualmente pueden adoptar medidas para prevenir el abuso de los derechos de propiedad intelectual (artículo 8.1 y 8.2 del Acuerdo).
Conviene tener en cuenta que, en los países que quedan obligados a introducir la protección por patente para los productos farmacéuticos como resultado del Acuerdo sobre los ADPIC, sólo se podrán patentar los productos para los cuales se haya solicitado la patente con posterioridad al 1 de enero de 1995. Esto quiere decir que otros productos (incluidos los que en esa fecha estaban ya patentados o en vías de serlo en otros países, o comercializados) permanecerán en el dominio público, a menos que la legislación nacional admita (como sucede en Brasil) la protección retroactiva de los productos en el llamado “pipeline”.
Dada la diversidad de los objetivos nacionales, no es sorprendente que los sistemas de patentes de los distintos países diverjan, en algunos casos de forma notable. No hay un “sistema de patentes” único. Además, las soluciones adoptadas en algunos países han variado a lo largo del tiempo. En el futuro es posible que sigan evolucionando para responder mejor a consideraciones de equidad y a la naturaleza de la innovación en las ”tecnologías de sistemas acumulativos”.
Los países tratan aspectos específicos de las patentes (incluidos los requisitos de elegibilidad, el alcance de la protección, las excepciones a los derechos exclusivos y las licencias obligatorias) de maneras muy diversas. A la hora de trazar su normativa propia sobre DPI, los responsables políticos y legisladores de los países en desarrollo deben reconocer que, incluso dentro del marco general de los tratados internacionales, tienen un margen considerable para diseñar y aplicar soluciones propias a cuestiones particulares. Cada país satisfará mejor sus necesidades si sabe aprovechar la variada experiencia de los sistemas nacionales de todo el mundo, lo cual quiere decir que es valioso un buen conocimiento del derecho comparado.
Algunos países, sobre todo desarrollados, han optado por sistemas jurídicos que confieren derechos de patente fuertes. Lo han hecho con el fin de proteger la corriente de ingresos que les produce su base tecnológica ya asentada y promover la inversión en innovación tecnológica. En esos países existe, no obstante, un debate considerable sobre cuáles son el nivel y alcance óptimos de la protección para fomentar la innovación sin restringir indebidamente la libre circulación de las ideas ni sofocar la competencia. En algunos países se manifiesta una preocupación creciente por las deficiencias del proceso de examen y la proliferación de patentes de baja calidad. Además, la teoría económica del derecho de patentes sigue siendo un área incierta, donde todavía se echan en falta un marco teórico robusto y evidencia empírica concluyente.
Lógicamente, es posible que los países menos adelantados en el aspecto tecnológico prefieran promover la transferencia de tecnologías necesarias para su desarrollo, y preservar y fortalecer la competencia para asegurarse el acceso a productos, servicios y tecnologías en las condiciones de mercado más favorables. Incluso en países que propugnan y practican la protección más fuerte para los DPI, las leyes nacionales suministran mecanismos compensadores que protegen contra el posible abuso de los poderes a que los DPI dan lugar: la provisión de licencias obligatorias es un ejemplo.
Al diseñar un sistema nacional de patentes, las autoridades responsables deben tener en cuenta cuestiones transversales como son la protección del medio ambiente y de la salud pública, el fomento de la competencia y de la transferencia de tecnología, la protección de los consumidores y el apoyo a los pequeños inventores del país, respetando al mismo tiempo los derechos de los inventores a ser recompensados por sus aportaciones al progreso técnico.
Además, se deberían considerar atentamente otras medidas reguladoras que afecten a la salud pública, por ejemplo las relativas a la aprobación y el registro de medicinas, con miras a desarrollar un marco jurídico coherente que facilite el acceso a las medicinas necesarias.
La protección de la salud pública es uno de los problemas más apremiantes en los países en desarrollo. Una gran parte de la población del mundo aún no tiene acceso a los medicamentos esenciales; en las regiones más pobres de África, por ejemplo, más del 50 por ciento de la población carece de él. Se calcula que 1.500 millones de personas no llegarán a cumplir sesenta años, y más de 880 millones de personas no disponen de atención sanitaria. De los más de 33 millones de personas seropositivas que hay en el mundo, el 95 por ciento viven en países en desarrollo, y en la mayoría de los casos no pueden costearse los medicamentos necesarios. Para hacer frente a esa dramática situación se necesita un planteamiento integrado de los problemas, profundamente interrelacionados, de las políticas nacionales de salud, farmacéutica y de patentes. Ninguna de esas políticas se puede trazar ni aplicar aisladamente.
MATERIA PATENTABLE
Productos
Cuando se inició la Ronda Uruguay de negociaciones comerciales, más de cincuenta países (entre ellos algunos desarrollados) no otorgaban protección de patente a los productos farmacéuticos. Algunos juzgaban necesaria esa desprotección para promover el acceso a los medicamentos a precios competitivos, mientras que otros la censuraban alegando que pone en peligro la innovación y priva injustamente a los inventores de los beneficios que generan sus aportaciones.
El Acuerdo sobre los ADPIC obliga a todos los Estados Miembros de la OMC a reconocer patentes en todos los campos de tecnología (artículo 27.1). Cuando esté plenamente en vigor, esta obligación habrá eliminado los diferentes planteamientos de la política de patentes que existieron antes. Interpretado al pie de la letra, el artículo 27.1 no permite excluir de la patentabilidad las medicinas en general, ni, cabe sostener, grupos específicos de medicinas. Según esta interpretación, los Estados Miembros de la OMC no podrían excluir de la patentabilidad ni siquiera los “medicamentos esenciales” enumerados por la Organización Mundial de la Salud (OMS).
El Acuerdo sobre los ADPIC admite dos excepciones en virtud de las cuales no sería imposible excluir los productos farmacéuticos de la patentabilidad, pero ninguna de las dos parece suficiente para justificar esa exclusión, salvo en circunstancias limitadas.
La primera es el orden público, una de las razones que el artículo 27.2 del Acuerdo reconoce para excluir de la patentabilidad. Dado que no existe una idea del orden público universalmente aceptada, los Estados Miembros disponen de cierta flexibilidad para definir las situaciones cubiertas, dependiendo de sus particulares valores sociales y culturales. El propio artículo 27.2 indica que el concepto no se circunscribe a las razones de “seguridad”; también se refiere a la protección de “la salud o la vida de las personas o de los animales” o “vegetales”, y se puede aplicar a invenciones que puedan ocasionar “daños graves al medio ambiente”.
El artículo 27.2 indica que la no patentabilidad por razones de orden público es admisible si se previene al mismo tiempo la explotación comercial de la invención. En otras palabras, quizá no sea posible declarar la no patentabilidad de una invención si al mismo tiempo se permite su distribución o venta.
No obstante, la situación podría ser distinta si los países en desarrollo de todo el mundo (o sus organizaciones regionales) decidieran colectivamente prohibir o suspender la patentabilidad de ciertos productos farmacéuticos por motivos de orden público. Una decisión de esa clase podría generar una nueva “práctica estatal” que los grupos especiales de la OMC tendrían que tener en cuenta. Si los motivos de semejante decisión fueran lo bastante poderosos para justificar al menos una ampliación temporal de la excepción por orden público más allá de sus referencias tradicionales, es concebible que también pudieran justificar una excepción a la norma de “no explotación comercial” contenida en el artículo 27.2, siempre que los productos en cuestión se distribuyeran con fines no lucrativos. Estas cuestiones son intrínsecamente especulativas, y hasta cierto punto contingentes respecto al sentido, que aún está por determinar, de las disposiciones de salvaguardia incluidas en el Acuerdo sobre los ADPIC (véanse los artículos 7 y 8).
Una segunda excepción que podría autorizar la exclusión de los productos farmacéuticos de la patentabilidad se encuentra en el artículo 8.1 del Acuerdo sobre los ADPIC, el que reconoce explícitamente el derecho de los Miembros de la OMC a adoptar medidas dictadas por razones de salud pública. Sin embargo, las medidas que se adopten deberán ser “necesarias” y compatibles con las restantes obligaciones que el Acuerdo establece.
La exigencia de “compatibilidad” puede permitir exclusiones de la patentabilidad en casos de clara emergencia de salud pública definida como tal por el gobierno nacional, más allá de las medidas ordinarias o cotidianas en materia de salud y nutrición. Los casos de emergencia podrían poner en marcha la aplicación de una prueba distinta de “incompatibilidad” (conforme a lo dispuesto en el artículo 8.1), o calificarse como situación desfavorable para “el bienestar social y económico” (según el artículo 7). En esa hipótesis, una suspensión o exclusión de la patentabilidad podría ir aparejada a, y justificarse por, una emergencia concreta. Una vez que cesara la emergencia se podría restaurar el requisito de patentabilidad en los términos del Acuerdo.
Es evidente que una consideración clave es el propósito que indujera a adoptar la exclusión de una materia. Si, por ejemplo, se pudiera obtener el mismo objetivo imponiendo las licencias obligatorias que autoriza el artículo 31, en la exclusión de la patentabilidad se podría ver un mero intento de circunvenir las condiciones previas de dicho artículo. Ahora bien, si las situaciones locales planteasen problemas excepcionales que mereciesen atención en aras del interés público, esos problemas también podrían justificar la suspensión o limitación de otros artículos, como el artículo 31, en favor de alguna exclusión no permanente de patentabilidad, si tal exclusión fuera necesaria para resolver el problema.
Una cuestión que acaso convendría explorar más a fondo es la de si una excepción a la patentabilidad se puede justificar al amparo de la excepción general del GATT a las disciplinas comerciales, cuando la excepción es necesaria para proteger la salud pública (artículo XX(b)).
Este último artículo reconoce la importancia de que las naciones soberanas puedan promover los intereses de la salud de su población, aun cuando ello sea contrario a sus compromisos generales bajo los acuerdos de la OMC. Sin embargo, hasta ahora el artículo XX(b) se ha interpretado y aplicado de manera bastante restringida en la jurisprudencia del GATT/OMC. y es dudoso que el artículo XX(b) del GATT sea de aplicación en el contexto del Acuerdo sobre los ADPIC. En opinión de un Grupo Especial, el Acuerdo sobre los ADPIC goza de una condición jurídica autónoma, sui generis, dentro de la OMC, a pesar de ser “parte integrante del sistema de la OMC, el cual se basa en la experiencia adquirida durante casi medio siglo” en el marco del GATT.
En resumen, excluir de plano la patentabilidad de los productos farmacéuticos (ni siquiera limitada a la categoría de medicamentos esenciales) parece no ser una opción viable con arreglo al vigente Acuerdo sobre los ADPIC. La admisibilidad de excepciones fundadas en razones de orden público dependerá de la interpretación que se haga tanto del artículo 27.2 como de los artículos 7 y 8, pero no parece una base prometedora para excluir la patentabilidad. Las exclusiones orientadas a atender emergencias concretas de la salud pública, sobre todo por un período de tiempo limitado, podrían ser justificables si fueran parte necesaria de una estrategia global para hacer frente a la emergencia.
Sustancias existentes en la naturaleza
Algunos productos farmacéuticos están basados o consisten en materias biológicas. A esta categoría pertenecen los compuestos extraídos de plantas y algas, así como las proteínas humanas obtenidas por extracción o mediante técnicas de ingeniería genética (p. ej., el interferón, la eritropoyetina, la hormona del crecimiento). Las plantas en particular son una fuente importante de medicinas.
El que los materiales biológicos sean patentables depende en buena medida de que califiquen como “invenciones” (y por lo tanto materia patentable) o “descubrimientos” (materia no patentable). Distintas tradiciones del derecho de patentes dan distinto tratamiento a esta cuestión.
Si la filosofía que subyace a la ley de patentes es que una protección amplia puede fomentar la actividad inventiva, las excepciones para la materia biológica pueden parecer innecesarias, e incluso contraproducentes. Además, algunos países en desarrollo pueden temer que excluir la patentabilidad de sustancias existentes en la naturaleza signifique un obstáculo para la inversión en ciertas actividades locales, entre ellas algunas que podrían llevar a patentar productos derivados del conocimiento tradicional o de conocimientos prácticos propios del lugar. De todos modos, la importancia de un desincentivo de esa clase dependería de la capacidad industrial local y de la existencia de leyes que suministren formas de protección alternativas, incluidas leyes sobre modelos de utilidad o para proteger know-how no secreto.
Los países que cuentan con capacidades escasas de investigación, y aquellos que priorizan la asequibilidad y el acceso a los medicamentos, pueden preferir un enfoque distinto, que trate de buscar limitaciones a la patentabilidad de las sustancias que existen en la naturaleza. Análogamente, los países que consideran que la patentabilidad de tales sustancias atenta contra valores culturales y éticos fundamentales pueden querer restringir la patentabilidad de las materias biológicas. La posibilidad de hacerlo estará, sin embargo, limitada por las disposiciones del Acuerdo sobre los ADPIC que establecen la patentabilidad de los microorganismos y de los procedimientos no biológicos o microbiológicos para la producción de plantas o animales (artículo 27.3.b).
Las legislaciones nacionales varían considerablemente en cuanto a la clasificación de las materias biológicas como invenciones o descubrimientos. En algunas jurisdicciones (p. ej., Estados Unidos), una forma aislada o purificada de un producto natural, incluidos los genes, es patentable. La Directiva europea sobre invenciones biotecnológicas (núm. 96/9/EC, de 11 de marzo de 1996) adopta un enfoque similar. La Directiva, que es esencialmente declaratoria del derecho que hace tiempo rige en Europa, establece que “la materia biológica” y las sustancias aisladas del medio natural (tales como antibióticos nuevos) se considerarán patentables.
El Acuerdo sobre los ADPIC no define qué constituye una “invención”; sólo especifica los requisitos que una invención debe cumplir para ser patentable. Esto deja a los Estados Miembros una considerable libertad para determinar a qué se debe considerar invención, y para excluir de la patentabilidad cualquier sustancia que exista en la naturaleza. En particular, las moléculas del ADN se pueden considerar elementos de construcción de la naturaleza, que deberían estar disponibles para uso de la comunidad científica y para cualquier aplicación productiva.
Usos
Las patentes farmacéuticas rara vez se refieren a nuevas entidades químicas, esto es, a ingredientes activos que representen una aportación inédita al conjunto de productos disponibles para su uso en medicina. Gran número de patentes farmacéuticas protegen procedimientos de fabricación, formulaciones, sistemas de administración y nuevos usos de un producto conocido.
Métodos de tratamiento y de diagnóstico
Los países en desarrollo pueden considerar la exclusión de la patentabilidad de los métodos de diagnóstico, terapéuticos y quirúrgicos para el tratamiento de personas o animales. La mayoría de los países no conceden patentes sobre tales métodos, debido a razones éticas o a la dificultad de controlar en la práctica su observancia. Además, un método que se aplica al cuerpo humano no es susceptible de aplicación industrial y, por lo tanto, no cumple con uno de los requisitos clave de patentabilidad de la mayoría de las legislaciones sobre patentes. Sin embargo, en los Estados Unidos la práctica en este terreno favorece cada vez más la concesión de patentes sobre métodos terapéuticos si éstos satisfacen la definición de procedimiento y las restantes condiciones de elegibilidad.
El artículo 27.3.a del Acuerdo sobre los ADPIC autoriza explícitamente a los Miembros a no conceder patentes sobre los métodos de tratamiento terapéutico, quirúrgico y de diagnóstico.
Medicinas tradicionales
La medicina tradicional basada en el uso de productos naturales y en los conocimientos que poseen las comunidades indígenas y locales tiene gran importancia en los sistemas de atención sanitaria de muchos países en desarrollo. Se ha calculado que en la medicina indígena se utilizan unas 7.500 especies vegetales, muchas de las cuales, como el añil, tienen múltiples aplicaciones. La posibilidad de proteger mediante patentes la medicina tradicional se tropieza con dos obstáculos principales. Primero, el requisito de novedad generalmente impedirá patentar tales productos. Segundo, decisiones políticas encaminadas a mejorar el acceso a las medicinas incluido un enfoque limitativo de la patentabilidad de los productos naturales y de los usos de productos existentes, así como requisitos de patentabilidad estrictos pueden llevar a la exclusión de la protección para la mayoría de los productos de la medicina tradicional.
Además, la protección de la medicina tradicional mediante patentes nacionales no eliminará el riesgo de ”biopiratería”. Dado que la concesión de patentes depende de cada legislación nacional, la no patentabilidad en un país no significa que el conocimiento tradicional no pueda ser patentado en otro país sin la autorización de las comunidades que desarrollaron o poseían ese conocimiento. En estos casos puede ser necesario reclamar la anulación de la patente indebidamente concedida en el país extranjero.
Se han hecho muchas propuestas para proteger el conocimiento tradicional (incluido el de uso medicinal) mediante un régimen sui generis. Es el caso, por ejemplo, de las propuestas de reconocimiento de ”derechos intelectuales tribales”, ”comunales” o ”comunitarios”, y de ”derechos sobre recursos tradicionales”, entre otros. El establecimiento de un régimen de esa clase no atentaría contra el Acuerdo sobre los ADPIC, toda vez que su efecto no sería restringir el alcance de la protección de la propiedad intelectual sino ampliarlo. Aparte de eso, el establecimiento de un régimen especial quedaría fuera del campo de aplicación del Acuerdo sobre los ADPIC, el que sólo afecta las categorías de derechos de propiedad intelectual que se especifican en su artículo 2.
Otros enfoques ajenos a la esfera de la propiedad intelectual pueden servir también para promover el uso de los conocimientos tradicionales para el cuidado preventivo y curativo de la salud, o impedir su apropiación no autorizada por investigadores o empresas de países extranjeros. La ley 8423 (1997) de Filipinas, por ejemplo, persigue ”acelerar el desarrollo de la atención sanitaria tradicional y alternativa” mejorando la fabricación, el control de calidad y la comercialización de medicinas tradicionales. El Perú aprobó una ley en julio de 1999 que prohibe la exportación sin valor añadido de algunas especies botánicas con propiedades curativas conocidas que habían llegado a ser objeto de extracción masiva por parte de laboratorios extranjeros. La ley cubre las dos plantas medicinales más conocidas de la farmacopea indígena del Perú, la uña de gato y la maca, y los legisladores han estudiado ampliar la norma para dar cabida a otros productos (yacón y parapara).
ALCANCE DE LAS REIVINDICACIONES
Las reivindicaciones de las patentes definen los derechos del inventor. El alcance de las reivindicaciones determina hasta dónde llega la protección del monopolio del inventor, y por lo tanto es un aspecto importante que hay que considerar a la hora de diseñar y aplicar las leyes de patentes nacionales. Esta cuestión es particularmente pertinente en el caso de las invenciones relacionadas con la salud, debido a las prácticas de concesión de patentes que prevalecen en este ámbito. Recientemente los expertos han advertido que patentes demasiado amplias en el campo de la biotecnología podrían excluir importantes herramientas de investigación del dominio público y cerrar áreas enteras a ulteriores investigaciones.66 También se ha cuestionado la protección amplia que a veces se otorga a invenciones relacionadas con productos farmacéuticos.
Las reivindicaciones de una patente consisten esencialmente en una definición de la invención redactada en un único enunciado, donde debe explicitarse sin ambigüedad la aportación técnica hecha por el inventor. El alcance de la protección que otorga la patente, y, en consecuencia, el margen que queda para la investigación independiente y la competencia de terceros, queda determinado por la redacción empleada en las reivindicaciones. El cómo se describa un producto y la cobertura de la patente son aspectos de particular importancia. En lo que sigue se dan ejemplos de algunas de las posibles formas y cobertura de las reivindicaciones de patentes de producto.
Un producto químico generalmente se puede describir en términos estructurales, especificando, por ejemplo, su composición química. Este tipo de descripción es el que ofrece la manera más segura de delimitar el alcance de la protección.
Algunos países aceptan, bajo ciertas condiciones, las reivindicaciones funcionales que describen la invención en términos de lo que hace más que de lo que es. Las reivindicaciones de esa clase pueden dar lugar a una cobertura extraordinariamente amplia, toda vez que confieren derechos exclusivos sobre cualquier medio que sea adecuado para lograr las funciones reivindicadas, es decir, protegen todas las maneras de resolver un problema.
Otra forma de reivindicación es la llamada reivindicación de producto por procedimiento, en la que el producto aparece caracterizado por el procedimiento que se emplea para obtenerlo, y no por sus elementos ni su estructura. Estas reivindicaciones son particularmente aplicables a los productos biológicos que no se pueden describir en términos de su estructura o composición (por ejemplo, en el caso de una macromolécula segregada por un microorganismo). La Oficina Europea de Patentes las acepta únicamente si el producto en sí es nuevo e inventivo, y por lo tanto patentable.
Las reivindicaciones de uso no protegen el producto sino el uso que se hace de él. Una infracción de una reivindicación de uso sólo se puede producir cuando el producto se prepara o se vende para el uso específico reivindicado en la patente.
En lo que concierne a la cobertura, las reivindicaciones pueden ser más o menos claras y precisas. Una reivindicación puede referirse a un compuesto bien definido de valor terapéutico. Es frecuente, sin embargo, que en los sectores químico y farmacéutico se redacten las reivindicaciones de una manera que cubre cientos e incluso miles de compuestos. Ése es el resultado, por ejemplo, de describir una familia de compuestos químicos enunciando el núcleo estructural común de todos los miembros que tienen un sustituyente variable.
Las legislaciones nacionales, incluidas las de los países desarrollados, tratan estas cuestiones de maneras muy diversas. Las reivindicaciones funcionales han sido generalmente admitidas en los Estados Unidos, aunque se ha condenado el empleo de una terminología funcional genérica que pueda impedir nuevos avances de investigación y desarrollo. La Oficina Europea de Patentes (OEP), por su parte, sólo acepta las reivindicaciones funcionales cuando no hay otra manera más precisa de describir la invención. Las reivindicaciones de ”producto por procedimiento” suelen ser admitidas por la OEP y algunos países europeos sólo si es imposible definir un producto por sus características estructurales, y si el producto obtenible es nuevo e inventivo. Bajo las reivindicaciones de ”producto por procedimiento”, la protección suele alcanzar solamente a un producto obtenido mediante el procedimiento reivindicado; por lo tanto, el mismo producto obtenido por otro procedimiento no significaría infracción de una reivindicación existente.
La aceptación de reivindicaciones no estructurales y de amplia cobertura ensancha el dominio sujeto al control de los titulares de patentes. Las reivindicaciones genéricas pueden tener un impacto negativo en la investigación y bloquear indebidamente la competencia. También es probable que originen muchos litigios, que en última instancia acrecentarían los costos para las compañías y los consumidores. Reduciendo el alcance de las patentes mediante la aplicación de normas estrictas a los requisitos de descripción de las reivindicaciones y su cobertura se abre más margen a la innovación y la competencia. Desde una perspectiva de salud pública, es necesario buscar un equilibrio adecuado en esta materia.
El Acuerdo sobre los ADPIC guarda absoluto silencio sobre estas materias. No hay nada en dicho Acuerdo que obligue a los Miembros a admitir reivindicaciones funcionales ni de los otros tipos descritos supra. Siempre que no exista discriminación basada en el campo de la tecnología, el Acuerdo otorga plena libertad a los Miembros para determinar la forma y los límites de las reivindicaciones admisibles. Cualquier Miembro de la OMC puede exigir que, siempre que sea posible, una invención de producto se defina con precisión en términos de su composición o estructura específica, particularmente en el campo de las sustancias químicas, con miras a evitar las reivindicaciones excesivamente genéricas y asegurar la practicabilidad de la invención. Esta exigencia puede ser particularmente útil para acrecentar el papel de la documentación de patentes como fuente de información, y facilitar la negociación de licencias contractuales y el uso efectivo de las invenciones patentadas.
Los reglamentos de aplicación de la ley de patentes pueden asimismo contener instrucciones específicas para las reivindicaciones correspondientes a diferentes campos de la tecnología, tales como los productos químicos o las invenciones digitales y mecánicas, que tengan en cuenta las características de cada campo.
REQUISITOS DE PATENTABILIDAD
Para poder obtener una patente, un inventor debe acreditar que su invención es nueva, manifiesta ”actividad inventiva” (es decir, no es obvia), y es susceptible de aplicación industrial.
La manera en que se definan y apliquen esos criterios es un determinante crucial del caudal de conocimiento que se sustrae del dominio público. Esta cuestión tiene una importancia decisiva para los productos farmacéuticos. Con frecuencia, el registro de un gran número de patentes sobre composiciones farmacéuticas, usos terapéuticos, polimorfos, procedimientos y/o formas de administración relacionados con un ingrediente activo permite a la compañía propietaria alzar una barrera elevada contra la competencia. Esas patentes (secundarias), si se aplican agresivamente mediante litigios “estratégic os”, o incluso “ficticios”, como instrumento para desalentar la competencia por parte de compañías locales, pueden expandir indebidamente el poder de mercado que otorgó la patente original. Tales abusos pueden alcanzar particular gravedad en países en desarrollo donde haya una tradición escasa o nula de controlar tales prácticas mediante una normativa antimonopolio.
No es fácil revocar patentes excesivamente genéricas o secundarias. Una vez que ha sido concedida, la patente se presume válida. Recae sobre la parte que la cuestiona la carga de probar que su otorgamiento fue indebido. Los consumidores, sobre todo en los países en desarrollo, rara vez poseen los recursos necesarios para impugnar las patentes excesivamente genéricas, aunque sufragan su costo en forma de precios más altos y menor acceso a los productos patentados.
La fuerte competencia interempresarial que existe en el sector farmacéutico ha dado origen a muchas oposiciones a patentes farmacéuticas por competidores afectados. Pero las empresas más pequeñas de genéricos de los países en desarrollo muchas veces carecen de recursos para iniciar acciones tan costosas. Además, la ola de fusiones y adquisiciones que ha tenido lugar durante la década de 1990 ha reducido espectacularmente el número de los actores principales y acentuado la estructura oligopolista del sector. Esta tendencia hace aún más importante administrar el sistema de patentes con miras a proteger a los competidores y al público, frente a las restricciones derivadas de patentes que se conceden bajo criterios de patentabilidad demasiado flexibles.
La flexibilidad o el rigor en la aplicación de los criterios de patentabilidad pueden variar de unos países a otros y a lo largo del tiempo. La interpretación y aplicación correctas de los criterios de patentabilidad son vitales para equilibrar los intereses públicos y privados, así como para evitar excesos que socavan la credibilidad del sistema de patentes.
Los estándares de novedad y actividad inventiva determinan hasta qué punto prevalece la libre competencia. Es comprensible que los países tecnológicamente adelantados, que invierten una parte sustancial de su Producto Nacional Bruto (PNB) en investigación y desarrollo, propicien normas de novedad permisivas y niveles bajos de actividad inventiva. Sin embargo, incluso esas políticas son cada día más controvertidas, en vista de la importancia de la innovación incremental en algunos sectores y del creciente número de patentes que protegen trivialidades. A propósito de esto se ha demostrado que una exigencia más alta de actividad inventiva puede acrecentar el valor de las patentes, porque las patentes concedidas con arreglo a un estándar más estricto son más fuertes y menos vulnerables a la oposición de competidores. En algunas industrias, ese efecto compensa con creces cualesquiera efectos de contar con menos patentes.
Los países tecnológicamente menos adelantados pueden preferir establecer exigencias más altas de novedad y actividad inventiva para preservar y robustecer la competencia sin vulnerar las normas mínimas internacionales. Con ello no harían sino seguir los pasos de muchos de los países adelantados de hoy, que adoptaron políticas semejantes cuando ellos mismos estaban en proceso de desarrollo.
Los responsables políticos deberían ser conscientes de que puede haber relaciones sutiles entre la novedad y la actividad inventiva. Por ejemplo, en la legislación tradicional sobre patentes de los Estados Unidos (sobre todo antes de que en 1982 se creara el Tribunal de Apelaciones del Circuito Federal), la exigencia de no obviedad era tan alta que los tribunales adoptaban una actitud relativamente blanda y permisiva hacia la novedad. Hoy día, en que el estándar de no obviedad se sitúa muy bajo, esa tradición permisiva puede ser anticompetitiva y perjudicial para el proceso de innovación, en tanto permita la concesión de patentes sobre invenciones que no difieren sustancialmente del estado de la técnica.
Los países en desarrollo deberían asimismo tomar nota de que exigencias altas de novedad o actividad inventiva pueden obrar en contra de innovadores locales que no sean capaces de satisfacerlas. Una manera de solventar este problema es adoptar una ley sui generis que trate de aquellas invenciones “de menor rango” que no alcanzan los niveles de novedad o actividad inventiva requeridos para una patente. Ejemplos de la tradición europea incluyen leyes sui generis del diseño industrial (que protegen los diseños de aspecto exterior) y leyes de los modelos de utilidad que pueden proteger las invenciones “menores” en general. Sin embargo, estudios recientes proponen también que los países en desarrollo adopten leyes para la protección de conocimientos prácticos no patentables bajo un régimen de derechos no exclusivos. Esas leyes podrían estimular la innovación mediante una compensación pero sin un derecho exclusivo.
Novedad
El sistema de patentes se concibió para recompensar al inventor por sus aportaciones al caudal de conocimiento preexistente. Los criterios utilizados para definir lo que es nuevo, son determinantes clave de las posibles limitaciones al libre acceso y uso de conocimientos técnicos y productos que se encuentran en el dominio público. Cuanto más estrictos sean los requisitos de novedad y otros, menor será el número de solicitudes que desemboquen en la concesión de una patente.
El requisito de novedad sirve para establecer si la invención reivindicada no está contenida en el estado de la técnica. Se aplica antes de considerar la existencia de actividad inventiva.
En las leyes modernas de patentes el requisito de novedad suele basarse en una evaluación del estado de la técnica de ámbito universal, esto es, en cualquier parte del mundo. En general, la novedad se destruye por la divulgación anterior por escrito, la utilización anterior u otra forma de comunicación pública de la invención.
Dentro de ese marco, la definición legal y aplicación del requisito de novedad difiere significativamente de unos países a otros. En algunas jurisdicciones se aplica una norma flexible, que permite conceder gran número de patentes. Por ejemplo, en los Estados Unidos la divulgación que haya tenido lugar fuera de los Estados Unidos sólo será destructiva de la novedad si se ha hecho por escrito.
Las legislaciones y prácticas nacionales difieren en muchos otros aspectos importantes:
· En los Estados Unidos, por ejemplo, la destrucción de la novedad exige la divulgación completa en una única publicación, a pesar de que una persona experta haya podido deducir la invención sin esfuerzo de una combinación de publicaciones.
· En algunos casos, la divulgación puede no haber sido hecha expressis verbis en un escrito anterior, sino estar implícita en el mismo. Si se aplica un planteamiento ”fotográfico” de la novedad (es decir, sólo basado en información explícitamente divulgada), los equivalentes
a una invención implícitamente divulgada en el estado de la técnica pueden no ser suficientes para negar la patentabilidad. El resultado en esos casos puede ser patentar fragmentos del conocimiento existente (estado de la técnica). Ese resultado se puede evitar siguiendo la práctica de la Oficina Europea de Patentes, que considera las enseñanzas implícitas como divulgadas y parte del estado de la técnica.
· Otro aspecto que se deja a la legislación nacional es el de establecer si la novedad únicamente se destruiría cuando la anticipación permitiera ejecutar la invención, o si sería suficiente una mera divulgación del estado de la técnica: por ejemplo, si se hiciera y ensayara un compuesto aunque no estuvieran disponibles una descripción clara de sus propiedades ni un método para hacerlo.
Actividad Inventiva
Aunque sea novedosa, una invención no es patentable si sus enseñanzas técnicas son tales que hubieran o pudieran haber sido establecidas a su debido tiempo por una persona medianamente experta en el campo correspondiente. En la práctica de los Estados Unidos, por ejemplo, los tribunales que aplican la norma de no obviedad (equivalente de la actividad inventiva en los otros países) ejecutan una indagación de los hechos en tres etapas, examinando:
(1) el alcance y contenido del estado de la técnica a la que pertenece la invención;
(2) las diferencias entre el estado de la técnica y las reivindicaciones en cuestión;
(3) el nivel de conocimientos ordinarios en la técnica pertinente.
A continuación los tribunales realizan una determinación final de la no obviedad decidiendo si una persona de conocimientos ordinarios en la materia en cuestión, podría salvar las diferencias que se aprecian entre el estado de la técnica y las reivindicaciones en cuestión, a la vista del estado de la técnica pertinente. Aunque a veces sea difícil de aplicar, el requisito de actividad inventiva o no obviedad es crítico para evitar la concesión de patentes sobre trivialidades.
Con frecuencia se evalúa la actividad inventiva considerando el efecto “inesperado” o “sorprendente” de la invención reivindicada. Sin embargo, los tribunales estadounidenses rechazan actualmente este enfoque, haciendo hincapié en que las invenciones patentables pueden ser el resultado de una investigación laboriosa, de un lento tanteo o de un hallazgo fortuito.
La jurisprudencia de muchos países sostiene que no existe actividad inventiva siempre que, para una persona medianamente experta en el tema, fuera obvio ensayar una materia nueva con una probabilidad de éxito significativa. En Estados Unidos la existencia de actividad inventiva con relación a compuestos químicos se ha juzgado tomando en cuenta la similitud estructural entre los compuestos reivindicados y los comprendidos en el estado de la técnica, la sugerencia o motivación que hubiera en el estado de la técnica para hacer el nuevo compuesto, y la obviedad del método empleado para obtener el compuesto que se reivindica.
Como en el caso de la novedad, las legislaciones nacionales pueden ser más o menos estrictas a la hora de evaluar la actividad inventiva o la “no obviedad”. Además, dentro de cada sistema legal es posible que los tribunales eleven o rebajen el estándar de actividad inventiva en distintas épocas, como respuesta a las actitudes imperantes hacia la competencia, la percepción de la necesidad de proteger nuevas tecnologías (tales como programas de ordenador e invenciones biotecnológicas), y la disponibilidad (o ausencia) de formas alternativas de protección en la normativa sobre competencia desleal, modelos de utilidad u otras.
Para establecer la existencia de actividad inventiva suele ser necesario considerar no sólo el conocimiento que se desprende de un único documento anterior, sino también el conocimiento combinado de la literatura existente, la documentación de patentes y otros componentes del estado de la técnica. Sin embargo, la práctica actual de los Estados Unidos es contraria a ese planteamiento, sosteniendo que “a materia objeto de una reivindicación no es obvia en virtud del estado de la técnica a menos que en el estado de la técnica exista alguna sugerencia o enseñanza específica que apunte hacia ella”.
En el campo químico y farmacéutico es frecuente que exista una relación estructural estrecha entre un compuesto que se reivindica como nuevo e inventivo y compuestos conocidos, tales como sales de ácidos, bases, isómeros y homólogos. En esos casos a menudo se puede considerar obvio ensayar el compuesto nuevo, de lo cual se sigue su no patentabilidad. La Oficina Europea de Patentes, por ejemplo, ha adoptado la postura de que el hecho de que ciertas ventajas fueran previsibles hacían obvio preparar un compuesto nuevo. En Estados Unidos, por el contrario, la presencia de una ventaja previsible no se considera suficiente para excluir la patentabilidad.
El Acuerdo sobre los ADPIC no desciende a detalles sobre la actividad inventiva. Su artículo 27.1 establece que podrán concederse patentes para proteger las invenciones que “entrañen una actividad inventiva”, y en nota al pie autoriza a los Estados Miembros a interpretar la expresión “actividad inventiva” como sinónimo de “no evidente”.
No hay actualmente bases para armonizar el estándar de actividad inventiva/no obviedad. Esto anima a pensar que los países en desarrollo harían bien en celebrar consultas y coordinar sus actuaciones sobre esta cuestión, posiblemente a través de sus organizaciones regionales.
Aplicación Industrial
El tercer criterio de patentabilidad se refiere a la aplicación industrial de la invención. En todo el mundo las leyes de patentes se ordenan a proteger las soluciones técnicas para un problema dado, no el conocimiento abstracto.
La aplicación de este criterio a las invenciones relacionadas con la salud es particularmente importante en lo que concierne a aquellas invenciones que consisten en usos de un producto, ya que los usos de invenciones relacionadas con la salud se pueden considerar como métodos de tratamiento del cuerpo humano, carentes de aplicación industrial y por lo tanto no patentables.
El Acuerdo sobre los ADPIC no define el concepto de aplicación industrial, y por lo tanto deja un margen de flexibilidad considerable a los países.
ESTÁNDARES DE PATENTABILIDAD
Si bien el art. 27.1 citado prescribe cuáles son los requisitos de patentabilidad, él permite a los miembros definir los alcances y condiciones para evaluar la novedad, actividad inventiva (o no obviedad) y aplicabilidad industrial (o utilidad). De hecho, conviven actualmente muy diversos criterios en estas materias que surgen tanto de la propia legislación como de las prácticas de las oficinas de patentes. Estas diferencias dejan margen para que los países en desarrollo establezcan, dentro de ciertos límites, sus políticas de patentabilidad conforme a sus condiciones y propios objetivos en materia de desarrollo industrial e innovación sin tener que subordinarse a políticas y prácticas desarrolladas en países más industrializados.
Por ejemplo, la novedad en los Estados Unidos sólo es destruida por una publicación escrita en el exterior; otros medios de divulgación tienen efecto destructivo de aquélla sólo cuando la divulgación tuvo lugar en el territorio de ese país. Este criterio contrasta con el de novedad universal empleado en Europa y en la mayoría de los países en el mundo. Igualmente, es sabido que el requisito de "utilidad" aplicado en los Estados Unidos expande la patentabilidad mucho más allá que el de aplicabilidad industrial. Aquél permite la protección de desarrollos carentes de un "efecto técnico", como en el caso de los programas de computación y los métodos de negocios, respecto de los que se han concedido miles de patentes en ese país.
Por otra parte, los estándares aplicados en la práctica para juzgar la novedad y altura inventiva o no obviedad divergen. Así, en algunos países la divulgación puede no haber sido hecha expressis verbis en una publicación anterior sino estar implícita en la misma. Algunas oficinas aplican un criterio "fotográfico" para evaluar la novedad (es decir, sólo basado en información explícitamente divulgada), de lo que resulta que los equivalentes a una invención implícitamente divulgada en el estado de la técnica pueden no ser suficientes para negar la patentabilidad. Este resultado se puede evitar considerando las enseñanzas implícitas como divulgadas y parte del estado de la técnica .
En relación con la actividad inventiva (o no obviedad) las diferencias son probablemente mayores. En algunos países se evalúa la actividad inventiva considerando el efecto "inesperado" o "sorprendente" de la invención reivindicada. Sin embargo, los tribunales estadounidenses rechazan actualmente este enfoque haciendo hincapié en que las invenciones patentables pueden ser el resultado de una investigación laboriosa, de un lento tanteo o de un hallazgo fortuito. Asimismo, la jurisprudencia de muchos países sostiene que no existe actividad inventiva cuando para una persona medianamente experta en el tema fuera obvio ensayar una materia nueva con una probabilidad de éxito significativa. En los Estados Unidos la existencia de actividad inventiva con relación a compuestos químicos se ha juzgado tomando en cuenta la similitud estructural entre los compuestos reivindicados y los comprendidos en el estado de la técnica, la sugerencia o motivación que hubiera en el estado de la técnica para hacer el nuevo compuesto y la obviedad del método empleado para obtener el compuesto que se reivindica.
La determinación de los estándares de novedad y actividad inventiva tiene gran importancia económica y social: ellos fijan hasta qué punto prevalece la libre competencia. Dada la importancia de la innovación incremental en algunos sectores, un bajo estándar de patentabilidad conduce a la proliferación de patentes, incluso sobre desarrollos triviales. La crítica es particularmente acentuada en los Estados Unidos, donde las academias nacionales de ciencias, haciéndose eco de las críticas de numerosos académicos y sectores de la industria, han iniciado un examen detenido del problema de calidad y cobertura de las patentes concedidas, en el orden ya de 160.000 a 180.000 por año.
Diversos estudiosos recomiendan mayor rigor en los requisitos para evitar la protección de desarrollos que distorsionan la finalidad del sistema de patentes y obstaculizan la competencia. Se ha demostrado que una exigencia más alta de actividad inventiva puede acrecentar el valor de las patentes porque las patentes concedidas con arreglo a un estándar más estricto son más fuertes y menos vulnerables a la oposición de competidores. En algunas industrias eso compensa con creces cualesquiera efectos de contar con menos patentes.
Dado el efecto de una política expansiva de patentabilidad (basada en bajos estándares de examen sustantivo y amplia cobertura de las reivindicaciones) sobre la competencia, la difusión de tecnologías y el consumidor, el Banco Mundial ha señalado que los países en desarrollo "...could set high standards for the inventive step, thereby preventing routine discoveries from being patented. Regarding patent scope, it is sensible to exercise strict claims and discourage multiple claims in patent applications".
En forma similar, la Comisión sobre Derechos de Propiedad Intelectual (CIPR.), creada por el gobierno británico en 2001, recomendó lo siguiente: "Developing countries should, within the constraints of international and bilateral obligations, provide a procompetitive patent system that limits the scope of subject matter that can be patented; applies strict standards of patentability; facilitates competition, includes extensive safeguards against abuses of patent tights, and encourage local innovation".
PATENTABILIDAD FARMACÉUTICA
La laxitud o rigor con que se evalúan las solicitudes de patentes tienen particular importancia en el sector farmacéutico. Aun cuando una patente obtenida puede ser débil y cuestionable, la experiencia muestra que las empresas titulares pueden hacerla valer agresiva y eficazmente contra posibles competidores locales, especialmente cuando (como sucede en varios países de América Latina actualmente) los jueces conceden con generosidad medidas precautorias inaudita parte que excluyen del mercado al supuesto infractor hasta que pueda probarse (varios años más tarde) la invalidez de la patente o la ausencia de infracción.
De hecho, las grandes empresas del sector han generado gran capacidad no sólo para desarrollar inventos genuinos sino para obtener patentes sobre adiciones al conocimiento secundarias, que se usan para extender el monopolio sobre un producto o proceso, más allá de lo permitido por la patente original, lo que en Estados Unidos se conoce como evergreening. Esta práctica, cuestionada incluso en ese país, afecta directamente al consumidor y a la salud pública en la medida en que se excluyan la competencia y, con ello, el acceso a los medicamentos. Se ha observado, en tal sentido, que: "En materia referente a invenciones del campo de la industria farmacéutica, se debe tener mucho cuidado para asegurar que las patentes sólo sean otorgadas a aquellos desarrollos que constituyen una efectiva contribución al estado del arte, y a impedir las patentes sobre invenciones triviales" (Soto Vázquez, Ramón, Cárdenas y Espinosa, Rodrigo A., Parra Cervantes, Patricia y Cassaigne Hernández, Rocío, "Protección a la inventiva farmacéutica", Asociación Farmacéutica Mexicana, 2001, México, p. 52).
El examen de la patentabilidad en el campo farmacéutico plantea algunos problemas particulares. Entre ellos cabe mencionar los siguientes:
Polimorfismo
Algunos ingredientes terapéuticamente activos presentan formas polimorfas, esto es, pueden cristalizar en formas diversas, las cuales pueden poseer propiedades físico-químicas diferentes. Se ha hecho frecuente solicitar patentes independientes para tales formas, aun cuando ellas no tienen efectos terapéuticos diferentes. En algunos casos se ha comprobado que tales formas están comprendidas en el estado del arte. En el de la cimetidina, por ejemplo, alrededor de cinco años después de patentarla, Smith Kline & French obtuvo una nueva patente sobre un polimorfo (una particular forma cristalina de la molécula) que, de hecho, ya había sido descripta en la patente original. La vigencia de esta patente significaba dilatar el ingreso al mercado de productos genéricos por varios años. La patente en cuestión fue cuestionada -exitosamente- en los tribunales de varios países por sobre la base de que el polimorfo se obtenía inevitablemente aplicando el procedimiento ya reivindicado en la patente original.
Otro ejemplo es el caso de la ranetidina, en el que el titular de la patente obtuvo en los Estados Unidos la patente de un polimorfo que expiraba en 2002, mientras que la patente principal expiraba en 1995. Asimismo, Eli Lilly solicitó y obtuvo una patente en la Argentina sobre un polimorfo de la olanzapina que, según la empresa, tendría mayor estabilidad que el polimorfo conocido previamente. Demandadas dos empresas locales por la comercialización de olanzapina, se planteó la nulidad de la patente con base en que -como en el caso de la cimetidina antes mencionado- el polimorfo tardíamente patentado necesariamente se obtiene con el método descripto en la patente original del producto. Otro caso reciente en Gran Bretaña involucró un polimorfo de la paroxetina, bajo patente de Glaxo SmithKline, uno de los productos comercialmente más exitosos, con ventas globales anuales de más de 2000 millones de dólares.
PROCEDIMIENTOS ANÁLOGOS
Algunos países han permitido patentar procedimientos no novedosos (a veces denominados "procedimientos análogos") si el producto químico resultante es novedoso y manifiesta propiedades inesperadas. Así, "antes de que Alemania introdujera en 1968 la patente de producto químico, los llamados procedimientos por analogía o Analogieverfahren suplían en la práctica esa imposibilidad. Se caracterizaban por la protección del procedimiento que llevaba a la obtención de un producto nuevo aunque el conjunto de los materiales de partida fueran ya conocidos, siempre que el producto resultante fuera novedoso".
Esta admisión, sin embargo, ignora la distinción entre patente de producto y procedimiento y se funda en una ficción legal de novedad. En los países donde no se reconocía patente de producto farmacéutico dicha ficción podía conducir, en la práctica, a bloquear la comercialización de un producto en el dominio público. Un ejemplo de ello es el caso de la sal de besilato de amlodipina. Pfizer obtuvo en la Argentina la patente AR 242562 sobre un simple proceso de salificación, ampliamente conocido, que carece de novedad y altura inventiva. Con esta patente Pfizer procuró impedir la comercialización del producto (no patentado). La patente es actualmente objeto de un juicio de nulidad.
En Estados Unidos se ha sostenido que las reivindicaciones de "procedimientos análogos" no son patentables a menos que en sí mismos sean inventivos, pero se hizo una excepción para la biotecnología debido a que muchas "invenciones" biotecnológicas repiten procedimientos ya inventados en contextos ligeramente distintos. Este problema dio lugar a una enmienda de la ley en 1995, la que estableció que la reivindicación de un procedimiento biotecnológico no es obvia si implica materiales de partida nuevos y no obvios o produce un resultado nuevo y no obvio. Esta solución, pensada exclusivamente para la biotecnología, ha sido extendida por la jurisprudencia a otros campos de la tecnología , en el contexto de una jurisprudencia francamente favorable a la expansión de la patentabilidad.
COMPOSICIONES Y FORMULACIONES
Las composiciones pueden ser combinaciones de productos ya conocidos. Por ejemplo, en los Estados Unidos se han concedido patentes sobre la combinación de las formulaciones siguientes: aspirina 325 mg + carisoprodol 200 mg + fosfato de codeína 16 mg, con fecha de expiración 13/8/2002. Otro ejemplo es la patente sobre un tipo de formulación de diadosina ("ddl") (un fármaco importante para los pacientes seropositivos), concedida en Tailandia, sobre la base de una combinación del principio activo y un antiácido.
Las patentes sobre composiciones se refieren con frecuencia a un producto formulado que contiene un ingrediente activo y los aditivos convenientes. Por ejemplo, se han otorgado patentes por separado sobre las formas inyectable y oral de la ofloxacina, un medicamento de interés en el tratamiento de pacientes seropositivos. También existe una patente para uso tópico oftálmico.
Es común también que se patenten nuevas formas farmacéuticas de un producto, por ejemplo, en forma líquida cuando estaba disponible en estado sólido. Por ejemplo, la patente que SmithKline Beecham solicitó para paroxetina en estado líquido, o como sólido absorbido en o por otro sólido.
a) Isómeros ópticos
Un caso especial se plantea cuando un compuesto es un enantiómero ópticamente activo de un compuesto que antes sólo se conocía en forma racémica. Aunque algunas oficinas de patentes, como la Oficina Europea de Patentes europea, han dictaminado que tales enantiómeros se pueden considerar novedosos, se ha negado la existencia de actividad inventiva pues es obvio que en esos tipos de moléculas pueden existir formas ópticamente activas, y es habitual poner a prueba si uno u otro de los enantiómeros aislados es más activo que la mezcla de los dos ("mezcla racémica"). Hoy día se acepta en general que lo normal será que uno de los isómeros ópticos muestre una actividad mucho más alta que el otro, de suerte que es de esperar una actividad superior al menos en uno de los isómeros en comparación con el racemato.
b) Metabolitos activos
En algunos casos se pueden acumular patentes sobre un compuesto y sobre el metabolito activo que produce el efecto deseado en el organismo. Por ejemplo, en el caso de la terfenadina, que desde hacía muchos años se vendía como antihistamínico en el Reino Unido, el titular de la patente obtuvo una nueva patente sobre el metabolito activo e intentó bloquear la competencia en el mercado de la terfenadina luego de que había expirado la patente sobre ésta. Los tribunales consideraron que éste era un intento inaceptable de prolongar la protección de la patente.
Otro conflicto se presentó en el caso de la cefalosporina: "Zenith Laboratorios desarrolló una forma hemihidratada de cefalosporina, a la cual designó como CDC. Con anterioridad Bristol-Myers había obtenido diversas patentes sobre la cefalosporina, entre ellas una nueva forma cristalina monohidratada la cual posee características que la hacen particularmente adecuada para su presentación en la forma farmacéutica de cápsulas (US 4.504.657). La forma hemihidratada del CDC. de Zenith difiere estructuralmente de la forma monohidratada de Bristol-Myers en el número de moléculas de agua. En 1990 Zenith Laboratorios recibe la aprobación de la Food and Drug Administration (FDA.) para su CDC. y Bristol le inicia una demanda argumentando que está siendo infringida su patente '657, pues el compuesto hemihidratado de Zenith sufre una transformación metabólica en el estómago de los pacientes y se convierte precisamente en el compuesto monohidratado. Un compuesto que no infringe una patente antes de su ingestión por un paciente, se convierte, mediante una modificación metabólica llevada a cabo en el estómago del paciente, en un compuesto protegido por una patente en vigor".
c) Profármacos
Hay compuestos inactivos que al ser metabolizados en el organismo pueden producir un ingrediente terapéuticamente activo, llamado "profármaco". Los países deben determinar si la patente sobre el compuesto cubre al profármaco y hasta qué punto se debe permitir que las reivindicaciones relativas a ciertos compuestos se hagan extensivas a sus profármacos. Por ejemplo, en Gran Bretaña se dictaminó que las sales de la hetacilina (un aducto de la ampicilina), que se hidrolizan en el cuerpo inmediatamente formando ampicilina, infringían la patente de este último compuesto ya que la hetacilina era "una ampicilina disfrazada".
d) Sales
La protección concedida por una patente sobre un ingrediente activo puede extenderse en algunos casos mediante la protección de sales que poco o nada agregan en términos de altura inventiva, recurriendo a técnicas ampliamente difundidas. Por ejemplo, la paroxetina es conocida tanto en su forma de base como en la de sus sales de adición farmacéuticamente aceptables desde por lo menos 1977 por la publicación de la patente US 4007196, en la que hay referencias explícitas sobre la paroxetina base y sobre su maleato, en tanto el resto de sus sales de ácidos farmacéuticamente aceptables queda englobado en la fórmula general. Sin embargo, se obtuvieron diversas patentes sobre distintas formas de paroxetina base y de sus sales con distintos ácidos, en diversas formas (amorfa, cristalinas, hidratadas, anhidratos, solvatos, incluyendo distintos polimorfos de algunas de éstas). Recientemente se iniciaron varias acciones legales en la Argentina y en el Uruguay en relación con la comercialización de docetaxel, si bien sólo el trihidrato fue patentado (por Aventis), en tanto se encuentra en el dominio público en su forma anhidra. Las dos formas son igualmente eficaces desde un punto de vista terapéutico.
e) Patentes de selección
Una "patente de selección" es aquella bajo la cual un solo elemento o un pequeño segmento dentro de un grupo conocido es "seleccionado" y reivindicado independientemente, con base en rasgos particulares no mencionados en el grupo más extenso. Si el grupo de elementos extenso ya está patentado el titular de la patente puede servirse de la patente de selección para prolongar el plazo de protección más allá de la expiración de la patente original, al menos para el subconjunto seleccionado. Aunque en algunas jurisdicciones tales patentes se aceptan cuando los elementos seleccionados poseen una ventaja imprevista, las patentes de selección se han denegado cuando la supuesta ventaja era una propiedad compartida por todos o casi todos los elementos del grupo extenso. Alemania ha rechazado las invenciones de selección sosteniendo que la divulgación de un grupo de elementos, aunque sea extenso, es plenamente equivalente, a efectos de la actividad inventiva, a la divulgación de cada uno de los compuestos comprendidos en el grupo.
Un ejemplo de patente de selección es la familia de patentes entre las que se encuentra la británica GB 2078719 de la firma ICI., la que cubre compuestos que presentan actividad fungicida. Uno de los compuestos explícitamente descriptos y reivindicados es el 1,3-bis-(1,2,4)-triazol il-2-(2,4-diclorofenil)-propan-2- ol. Pfizer desarrolló con posterioridad un nuevo antifúngico que, si bien está cubierto genéricamente entre los productos reivindicados en la patente de ICI., no había sido especificado y que Pfizer describe y reivindica específicamente en una familia de patentes que invoca prioridades británicas de junio y octubre de 1981 y marzo de 1982. Se trata del antifúngico fluconazol, reivindicado en la EP 69442. Este producto únicamente se diferencia del previamente patentado en que posee dos átomos de flúor en lugar de los de cloro sobre el anillo bencénico.
LA "SEGUNDA INDICACIÓN" FARMACÉUTICA
En algunas jurisdicciones se han adoptado normas especiales o prácticas administrativas y judiciales que han permitido proteger la segunda indicación de un producto farmacéutico conocido. Mas ésta no es una solución universal y no se aplica en el Comunidad Andina y otros países latinoamericanos.
En Europa una ficción legal de novedad autoriza a patentar un producto conocido para una primera indicación farmacéutica. Conforme al art. 54.5 del Convenio sobre la Patente Europea, la identificación de la primera indicación médica de un producto conocido puede bastar para patentar el producto. De esta forma se neutraliza la prohibición de patentabilidad de los métodos terapéuticos prevista también por el Convenio sobre la Patente Europea (art. 52.4). El problema es, en efecto, que la identificación del nuevo uso (farmacéutico) de un producto existente equivale a la de un nuevo método terapéutico. Domeij explica las dificultades con que se ha enfrentado el derecho europeo de patentes para abordar la patentabilidad de los nuevos usos farmacéuticos en los siguientes términos: "No hay una diferencia real entre las reivindicaciones de patentes relativas al uso de una sustancia y aquellas relativas a un procedimiento terapéutico: en ambos casos una nueva actividad médica es patentada, esto es, una nueva manera de usar uno o más productos desconocidos. De este modo, las dificultades en el derecho de patentes europeo en cuanto a la protección de una nueva indicación médica para una sustancia conocida se deben a la combinación del requisito de la novedad (que impide reivindicaciones de productos) y la prohibición de patentar procesos médicos (que impide reivindicaciones de uso)".
En los casos en que se descubre una segunda indicación farmacéutica para un producto que ya era objeto de uso farmacéutico se plantean esencialmente los mismos problemas: la estructura química del producto es conocida, y, por tanto, no novedosa, y el uso del producto equivale a un método terapéutico.
A pesar de que el Convenio sobre la Patente Europea sólo autorizó a la OEP. a salvar estas objeciones fundamentales con base en una excepción restringida aplicable a la primera indicación farmacéutica, tras una intensa controversia la OEP. extendió la patentabilidad también a la segunda indicación farmacéutica.
La solución europea se basó en la admisión de reivindicaciones para el segundo uso terapéutico de un producto farmacéutico modeladas como reivindicaciones de procedimiento y no de uso como tal, para evitar la prohibición de patentar métodos terapéuticos. La oficina de patentes europea aceptó segundas indicaciones sólo si las reivindicaciones se redactan según la "fórmula suiza", esto es, como una reivindicación sobre un procedimiento en la forma: "Uso de X en la fabricación de un medicamento para el tratamiento de Y" (G 1-6/83, OJ EPO. 1985, 64).
Para llegar a esta conclusión el Enlarged Board of Appeal de la OEP. consideró que la única distinción posible en el derecho de patentes es entre un producto y un método, esto es, entre un fenómeno físico y una actividad, y dedujo de esto que un nuevo uso médico de un producto conocido debe ser un método médico, por lo tanto no patentable debido a la prohibición de patentar tales métodos. A continuación resolvió que la novedad del uso descubierto para un producto farmacéutico ya conocido puede impregnar de novedad al procedimiento (no novedoso) para la elaboración del medicamento respectivo. Es decir, la decisión se fundó en una ficción de novedad consistente en transferir al procedimiento un atributo del que carece, argumentando lo siguiente: "Parece justificable por analogía derivar la novedad de los procedimientos que forman parte de la materia del tipo de reivindicación de uso ahora considerado del nuevo uso terapéutico de un medicamento y ello independientemente del hecho de si el uso farmacéutico del medicamento fuera ya conocido o no. Debe entenderse claramente que la aplicación de este especial enfoque de derivación de la novedad sólo puede ser aplicada a reivindicaciones del uso de sustancias o composiciones con la intención de ser usadas en un método referido en el art. 52 CEP.".
Mediante el atajo conceptual de "derivar" la novedad del nuevo uso en beneficio del procedimiento (no novedoso):
- se evita la objeción de no patentabilidad del uso farmacéutico, porque no se reivindica un uso como tal sino un procedimiento;
- se salva la falta de novedad del procedimiento transfiriéndole, mediante una ficción, la novedad del que sólo goza el nuevo uso.
En la Comunidad Andina, por ejemplo, el art. 16 de la decisión 486 establece: "Los productos o procedimientos ya patentados, comprendidos en el estado de la técnica, de conformidad con el art. 2 de la presente decisión, no serán objeto de nueva patente, por el simple hecho de aducirse un uso distinto al originalmente comprendido par la patente inicial".
En aplicación de esta norma, el Tribunal de Justicia de la Comunidad Andina resolvió en la "Acción de incumplimiento interpuesta por la Secretaría General de la Comunidad Andina contra la República del Perú" (proceso 89-AI- 2000, 28/9/2001): "Para este tribunal resulta claro a partir de esta disposición, que el legislador andino determina con la misma, una condición adicional a los requisitos fijados en los primeros artículos de la decisión 344, al excluir de la posibilidad de patentamiento a los productos o los procedimientos que gocen ya de la protección que contienen la patente... por el simple hecho de atribuirse un uso distinto al originalmente comprendido por la patente inicial.
"La prohibición o exclusión consagrada en el art. 16 en comentario, contiene como presupuestos básicos a juicio del organismo, primeramente, la determinación de que los productos o los procedimientos para los cuales se requiere la nueva protección de una patente, se encuentran ya amparados por igual derecho y, en consecuencia, se han ubicado en el estado de la técnica por haberse hecho accesibles al público”.
"Al tribunal le resulta claro, que sólo aquello que es nuevo puede ser protegido por una patente, principio incorporado al derecho comunitario seguramente con el objeto de incentivar la investigación; por lo que conceder protección del Estado a productos o procedimientos carentes de novedad, resultaría atentatorio tanto al propósito señalado como a la misma función social asignada al derecho de propiedad industrial... El simple hecho de atribuirse un uso distinto al originalmente comprendido por la patente inicial, debe ser necesariamente entendido como la consagración en el art. 16 de la decisión 344, del principio que no podrá reclamarse patente para usos distintos del invento o de la invención comprendidos y protegidos ya por la patente inicial o primigenia; regla prohibitiva para el otorgamiento de patentes de invención, que este tribunal considera como parte de los requisitos establecidos por la referida decisión".
LICENCIAS OBLIGATORIAS
La concesión de licencias obligatorias permite al gobierno dar derecho a que una compañía, una agencia gubernamental u otro interesado utilice una patente sin el consentimiento de su titular. Una licencia obligatoria debe ser concedida por una autoridad competente a una persona designada, la cual generalmente deberá compensar al titular de la patente mediante el pago de una remuneración. Las licencias obligatorias no niegan a los titulares de patentes el derecho de actuar contra terceros sin licencia.
La provisión de licencias obligatorias es un elemento crucial en una ley de patentes que tenga en cuenta las exigencias de la salud. Estas licencias pueden ser un instrumento importante para fomentar la competencia y hacer más asequibles los medicamentos, asegurando al mismo tiempo que el titular de la patente sea compensado por el uso de la invención. Sin embargo, la industria farmacéutica basada en la investigación se ha opuesto en general a la utilización de tales licencias, alegando que desincentivan la inversión y la I+D.
ARGENTINA
El informe sobre la situación de salud de Argentina elaborado por la Organización Panamericana de la Salud correspondiente al año 2003, indica que los precios de los medicamentos crecieron durante la última década muy por encima de la inflación. En este contexto, y tras la acentuación de la debacle económica y la crisis social, vastos sectores de la población se vieron imposibilitados del acceso a los medicamentos, sobre todo con la eliminación de la convertibilidad.
Vale aclarar que los medicamentos genéricos son aquellos que poseen la misma forma farmacéutica e igual composición cuali y cuantitativa que otro medicamento de referencia. Es decir, debe haber bioequivalencia entre uno y otro. Sólo pueden comercializarse una vez que caducó la patente original y se distribuyen con el nombre del principio activo, sin identificarse con una marca comercial.
A principios de 2002, para enfrentar la emergencia socioeconómica, el Ministerio de Salud impulsó una política nacional de medicamentos cuyos principales pilares fueron la prescripción de medicamentos por el nombre genérico de la droga y el Programa “Remediar”.
El objetivo de la ley de genéricos es promover la competencia por precios con vistas a mejorar la calidad de los medicamentos.
Así, los médicos y odontólogos se vieron inducidos a recetar por nombre genérico y los profesionales farmacéuticos fueron habilitados para ofrecer las distintas alternativas existentes en plaza para un mismo principio activo.
Según un artículo publicado en agosto de 2003 por la cartera sanitaria nacional, las medidas tuvieron una importante repercusión en el mercado de medicamentos. Aumentaron la competencia y permitieron que el precio promedio de los fármacos se mantuviera relativamente estable desde junio de 2002. La misma fuente indica que la estrategia posibilitó que “más de 14 millones de personas que antes no podían comprar los medicamentos, hoy puedan hacerlo”.
Sin embargo, tratar de implantar la ley de medicamentos genéricos desató una reacción inmediata de los grandes capitales de la industria farmacéutica. Pero como la política de medicamentos genéricos estaba sustentada en un concepto técnico- científico-médico muy fuerte, el proyecto siguió adelante. Las empresas dejaron de presionar y sumaron los medicamentos genéricos a su producción bajo marcas comerciales.
Sin embargo, al cuadro de situación planteado es necesario aportar otro dato: la modificación de la ley de patentes por la Cámara de Diputados de la Nación el 4 de diciembre de 2003. Según varios legisladores de la cámara baja las modificaciones introducidas a la legislación, en la práctica, implican que los capitales multinacionales del mercado de la industria farmacéutica sean los principales beneficiarios.
No es menor mencionar que los cambios se hicieron sobre la base del acuerdo alcanzado por Argentina y Estados Unidos en la Organización Mundial del Comercio (OMC), bajo la presidencia de Eduardo Duhalde.
Las modificaciones introducidas refieren a la inversión de la carga de prueba, patentamiento de microorganismos, patentes transitorias y protección de datos de pruebas contra un uso comercial ilegal. Es decir, amplía los derechos sobre las patentes -antes alcanzaba sólo a los productos- a todo el proceso de fabricación, y abre el juego, además, a la aplicación de medidas cautelares a favor de las empresas que tengan patente original sobre los productos.
A los inversores de capitales extranjeros les preocupa que le ley no se implemente en su totalidad. Quienes invierten cifras millonarias en investigación y desarrollo durante mucho tiempo necesitan que la propiedad intelectual se encuentre protegida durante todo el período de la patente y que no se introduzcan los genéricos hasta tanto no se haya cumplido el período de protección de la propiedad intelectual, porque ese es el verdadero sentido de la patente. También consideran que la protección a través de una buena ley de patentes proporciona la oportunidad para que se desarrolle un mercado de medicamentos genéricos. Estiman que su existencia se debe a las millonarias inversiones que hacen los laboratorios en investigación y desarrollo. Están a favor de los genéricos siempre y cuando se respeten dos cosas: los tiempos establecidos en la ley de patentes de cada país, y que el público tenga la seguridad de que esos genéricos tienen la misma calidad y ofrezcan los mismos resultados que los medicamentos de marca. Y eso no siempre sucede.
ANEXO
| Tribunal: | Corte Sup. |
| Fecha: | 27/01/1987 |
| Partes: | Baricalla de Cisilotto, María del Carmen v. Gobierno Nacional |
| Publicado: | JA 1987-II-331. |
MEDICAMENTOS - SALUD PUBLICA - Régimen de comercialización para productos medicinales - Ley 16463
Con nota de AUGUSTO M. MORELLO
OPINION DE PROCURADOR GENERAL DE LA NACIÓN
1. La actora interpuso la presente acción de amparo en representación de su hijo menor de edad, contra el Ministerio de Salud y Acción Social de la Nación, a fin de que se autorice el suministro del complejo crotoxina A y B en su faz de investigación a dicho menor, internado en el Hospital de Niños de esta ciudad con diagnóstico de neuroblastoma grado 4.
Fundó su pedido por la vía del amparo "dado que ,como es de público conocimiento, el Ministerio de Salud y Acción Social no autoriza el tratamiento ni suministra el complejo enzimático" lo cual, añade, configura un peligro inminente contra la vida de su hijo.
Al reducir el Poder Ejecutivo -dice- el número de pacientes autorizados a recibir el tratamiento experimental a 80, viene a vulnerar de manera arbitraria el derecho a la vida, la preservación de la misma, y la igualdad de todos los habitantes ante la ley, fundando, en definitiva, el derecho que invoca en las normas pertinentes de la ley 16986 y de la CN.
2. Tras recabar los informes del caso, el magistrado de 1ª instancia, a f. 212, desestimo la acción dando por reproducidos los argumentos hechos valer en una causa análoga. Ellos son: 1) que de las constancias obrantes, que refieren los pasos dados por la autoridad administrativa en redor de las investigaciones sobre la crotoxina, en modo alguno puede calificarse su proceder como ilegítimo o arbitrario; 2) que la decisión de continuar la investigación con el número de pacientes que a la fecha se encontraban en tratamiento, en modo alguno resulta irrazonable dada la etapa y las condiciones en que se encuentra la investigación, motivo por el que no puede reputarse violada la igualdad ante la ley; 3) que asimismo, "la administración de sustancias cuyas propiedades y características no han sido estudiadas y documentadas científicamente, no puede ser aceptada por las modernas sociedades".
3. Apelado ese pronunciamiento el recurso fue desestimado por el tribunal a quo, con base en las siguientes razones: a) el derecho a la vida y el derecho a la salud, quedan sujetos a lo que por la vía legislativa o administrativa se determine "en la medida de los recursos disponibles"; b) la decisión sobre la producción de la droga de que se trata "es por regla privativa de los poderes políticos"; c) la autorización de que la crotoxina sea usada como medicamento "está sujeta a facultades discrecionales de la administración" que en el sub lite no han sido ejercidas de modo discrecional; d) que dadas las limitaciones de disponibilidad de la sustancia "es razonable que se haya circunscripto la cantidad de pacientes".
4. Contra esta decisión la actora dedujo recurso extraordinario, el cual estimo que debe ser rechazado por haberse convertido en abstracto.
En efecto, V.E. tiene muy dicho que no corresponde pronunciamiento de la Corte cuando circunstancias sobrevinientes han tornado inoficioso decidir la cuestión materia de la litis (Fallos 306:157) y que las sentencias de la Corte deben atender a las circunstancias existentes al momento de la decisión, aunque ellas sean sobrevinientes a la interposición del recurso extraordinario (Fallos 306:1160).
Ello porque es de la esencia del Poder Judicial decidir colisiones efectivas de derechos, motivo por el cual no es propio de los jueces efectuar declaraciones generales o abstractas (Fallos 2:254; 12:372; 236:673, etc.).
En consecuencia, cabe advertir que el Ministerio de Salud y Acción Social -Secretaría de Salud-ha dictado, la resolución n. 47 de fecha 13 de octubre del corriente, que en su art. 2 dispone: "La producción, elaboración, comercialización, uso y aplicación en medicina humana del compuesto enzimático crotoxina A y B, se encuentran comprendidos en las previsiones del art. 19 inc. b ) y concs. ley 16463, debiendo hacerle saber esta circunstancia, en forma fehaciente. . .".
De resultas, por tanto, de esta nueva disposición deviene nítido que la materia de este pleito carece a la fecha de realidad efectiva, desde que por su intermedio se intentó la inclusión del hijo de la actora en los supuestos beneficios de un plan experimental que se ha venido a dejar sin efecto por el dictado de normas posteriores (conf. Fallos 292:375 y sus citas).
No obstante lo expuesto, atento a la índole particular de la cuestión litigiosa, y de modo escueto dada la abstracción de referencia en que ésta ha devenido, creo necesario destacar que, de todos modos, la actora carece de razones jurídicas formales y de fondo para pretender el acogimiento de su -desde ya- dramático y delicado reclamo.
Porque al margen de estos últimos ribetes, que sin duda no dejan de sacudir, con su carga emotiva, el problema de derecho que se presenta en el sub lite, lo rigurosamente cierto es que aquéllos no pueden torcer ni complicar la acertada solución jurídica de éste.
Y en tal sentido encuentro correcta la decisión de los jueces de la causa, e infundado el recurso federal del accionante, toda vez que éste se dedica a enfatizar las razones que expresó ante el a quo, mas no, como debía, a replicar los argumentos mediante los que el juzgador las desbaratase.
Si hay argumento, a mi criterio, basal, en la decisión recurrida, es el que concluye que el derecho a la salud, que invoca la actora está sujeto, como no puede ser de otra manera, a las reglamentaciones legislativas y administrativas pertinentes y en todos los casos, además, limitado por las posibilidades efectivas con que cuente el Estado.
El marco natural de desenvolvimiento de estas posibilidades y de aquellas reglamentaciones es diáfano y pertenece de lleno a la órbita de los otros poderes de gobierno, en cuyo ámbito sólo al Poder Judicial le compete, por virtud de su propio menester constitucional, el control de legalidad, de razonabilidad y de constitucionalidad.
Esto es que, con estricta referencia al sub exámine, la decisión de promover oficialmente la investigación en torno a los eventuales efectos curativos de una sustancia, los modos de concretarla, la determinación de los subsidios económicos para sostenerla y hasta la decisión en punto a los eventuales pacientes aceptados para favorecerla, son todos ellos temas de indubitable incumbencia exclusiva de los poderes administrativos y legislativos,sobre los que, a lo sumo, cabe con referencia a determinados aspectos un limitado control de los jueces, pero nunca, so pena de grave violación del orden constitucional, la pretensión de forzar por las vías procesales la intromisión del Poder Judicial en las funciones excluyentes de los otros poderes.
Así es bueno recordar aquí que V.E. tiene dicho con relación a otros supuestos específicos pero válidos como principios generales aplicables en la causa sub examine, que no se justifica la intervención de los jueces a fin de modificar la resolución de la administración en cuestiones que por su naturaleza les son propias (Fallos 301:291), que no es materia justiciable la revisión de la política administrativa, porque juegan apreciaciones que escapan, por su naturaleza, al poder de los jueces (Fallos 301:291), que no incumbe a los jueces en el ejercicio regular de sus atribuciones, sustituirse a los otros poderes del Estado en las funciones que le son propias (Fallos 270:169), que está vedado a los tribunales el juicio sobre el mero acierto o conveniencia de disposiciones adoptadas por los otros poderes en ejercicio de las facultades propias de ellos (Fallos 272:99; 277:25), y que, en definitiva, la misión más delicada de la justicia es la de saberse mantener dentro de la órbita de su jurisdicción, sin menoscabar las funciones que incumben a los otros poderes o jurisdicciones (Fallos 272:231).
Por último, también resulta necesario poner de resalto que si dicha intención de perseguir en la esfera judicial el dictado de decisiones que son propias y exclusivas de los otros órganos de gobierno no resulta jurídicamente admisible, menos aún lo es pretenderlo a través de la vía excepcional y sumarísima del amparo, desde que esta característica expeditiva y sumaria de la acción prevista en la ley 16986 no puede ser la idónea para, por principio, dirimir conflictos complejos donde precisamente debe ahondarse en tal complejidad a fin de no caer, por parte del poder jurisdiccional, en la invasión del campo de los restantes poderes.
Es obvio que en el triste problema de que aquí se trata, donde se apela a la solución urgente en razón de estar en juego en términos que se supone médicamente perentorios la vida del hijo menor de la actora, no pareciera que otra que no fuese la del amparo pudiese ser la senda procesal hábil para transitar el reclamo deducido, mas por riguroso que se presente a las conciencias legas debe concluirse que esta circunstancia vital y urgente no puede lograr de por sí la desarticulación del sabio mecanismo constitucional antes referido, que veda a los jueces suplir en las decisiones políticas legislativas a los funcionarios determinados para tales menesteres.
Por tanto, tampoco está demás recordar que V.E. tiene a su vez dicho que el recurso de amparo, de trámite sumarísimo, no procede en el supuesto de cuestiones opinables, que requieren debate y prueba (Fallos 271:165; 273:84; 274:186; 281:394), que asimismo no es la vía adecuada para enervar la validez de una decisión de autoridad competente, adoptada en ejercicio de atribuciones legales (Fallos 273:285; 274:365), ni es su razón de ser la de someter a la vigilancia judicial el desempeño de los funcionarios y organismos administrativos, controlando el acierto o la razonabilidad de la actividad de la autoridad administrativa, en tanto no medie arbitrariedad (Fallos 302:535).
Para el caso concreto, además, debe recordarse que también la Corte dijo que las decisiones en los juicios de amparo deben atender a la situación existente al momento de ser dictadas (Fallos 300:844) y que no es el procedimiento adecuado para discutir el reconocimiento de los derechos en abstracto (Fallos 270:367) .
Opino, en consecuencia, que el recurso debe ser rechazado. - Juan O. Gauna.
Buenos Aires, enero 27 de 1987.
Considerando:
1. Que la Sra. de Cisilotto, en representación de su hijo menor de edad, inició esta acción de amparo a fin de que el Estado Nacional (Ministerio de Salud Pública y Acción Social), le suministre el denominado complejo crotoxina A y B, en las dosis necesarias que requiere la enfermedad cancerosa que padece el menor. La demanda fue rechazada en 1ª instancia, y este pronunciamiento fue confirmado por la sala 3ª de la Cámara Nacional de Apelaciones en lo Contenciosoadministrativo Federal. Ello dio lugar al presente recurso extraordinario, que fue concedido.
2. Que, en primer lugar, es necesario determinar la pretensión en juego. En tal sentido, se observa que algunos pasajes del escrito de demanda traducen un reclamo de que el actor sea incluido en el ámbito de la resolución n. 522 del Ministerio citado, por la que se autorizó "la investigación clínica aplicaca del denominado complejo crotoxina A y B, en los enfermos actualmente sujetos a esa experiencia farmacológica. . ." (art. 1 , 25/07/1986). Desde ese ángulo es cierto, como lo señala el procurador general, que la causa se habría vuelto abstracta ya que tal investigación se dio por finalizada mediante la resolución n. 47 de ese ministerio (13 de octubre de ese mismo año, B.O. n. 26.016 del 16/10/1986). Empero, una interpretación integral de ese escrito, permite concluir que su objeto no se halla limitado al antes expuesto y, por ende, supeditado a la vigencia de la resolución n. 522 citada, sino que su alcance es mayor y comprensivo de la pretensión de que el Estado sea condenado a suministrar al actor el complejo mencionado. Luego, no es inoficioso decidir la causa sub exámine.
3. Que, sin embargo, lo expuesto no apareja la irrelevancia de la resolución n. 47 citada para resolver el litigio pues, como se verá, constituye aquélla un valioso elemento de juicio a tal propósito, máxime porque es doctrina permanente del tribunal que sus pronunciamientos deben atender a las circunstancias existentes al momento en que se los dicta, aunque ellas sean sobrevinientes a la interposición del recurso (Fallos 269:31; 292:140; 300:844, y sentencia dictada el 29/08/1986 in re Klein, Guillermo W.).
4. Que, en tales condiciones, es de señalar que el fundamento legal de la demanda no es otro que el derecho a la vida en cuanto comprensivo de la salud.
Con todo, si bien esta Corte ha declarado que el derecho a la vida es el primer derecho de la persona humana, que resulta reconocido y garantizado por la CN., es de la mayor importancia advertir la especial perspectiva de ese derecho.
En efecto, el derecho a la vida es invocado como fundamento por el cual el actor podría exigir, y el Estado estaría obligado a satisfacer, una prestación de salud consistente en suministrar determinada sustancia para lo cual debería, además, elaborarla previamente, por cuanto, como lo anuncia la resolución n. 47 citada, el Estado "no (la) posee ni produce".
No es la presente, por cierto, la oportunidad de referirse a todas las facetas del aludido derecho subjetivo.
Tampoco lo es la de estudiar si están reunidas todas las condiciones a que podría supeditarse la alegación válida de ese particular aspecto del derecho a la vida: gravedad de la situación; necesidad, insustituibilidad y eficacia del tratamiento; existencia de los medios necesarios para su prestación y el efecto que su empleo podría producir sobre la política general en materia de salud pública.
Sí lo es, por el contrario, la de analizar uno de esos recaudos pues sobre él versa la resolución n. 47 citada y, además, las conclusiones a que se arribarán son suficientes para juzgar el sub lite.
Resulta imprescindible reiterar que es ajeno a este debate todo lo concerniente a los alcances de la libertad de elección del tratamiento terapéutico por el paciente o por su médico. De lo que se tratará en esta sentencia, exclusivamente, es de analizar una de las condiciones mediante las cuales se le podría exigir al Estado, según nuestra Ley Fundamental, un tratamiento ya escogido, y si ese requisito ha sido o no acreditado.
5. Que, en tales condiciones, es a todas luces razonable afirmar que es condición inexcusable del ejercicio legítimo de ese derecho, que el tratamiento reclamado tenga eficiencia para el fin que lo motiva. En el caso, tal objetivo es el de combatir el cáncer.
6. Que las actividades de importación, exportación, producción, elaboración, fraccionamiento, comercialización o depósito en jurisdicción nacional o con destino al comercio interprovincial de las drogas, productos químicos, reactivos, formas farmacéuticas, medicamentos, elementos de diagnóstico y todo otro producto de uso y aplicación en la medicina humana, están sometidas a la ley 16463 -y a los reglamentos que en su consecuencia se dicten- y sólo pueden realizarse previa autorización y bajo control del Ministerio de Asistencia Social y Salud Pública (hoy Ministerio de Salud Pública y Acción Social), el que ejerce el poder de policía sanitaria referente a dichas actividades y se halla facultado para dictar las disposiciones reglamentarias o complementarias que sean necesarias para el cumplimiento de la finalidad del decreto 9763/1964, reglamentario de la ley 16463 (arts. 1 y 2 ley cit. y 40 decreto cit.).
Por otro lado, es ratio manifiesta de ambas normas, en lo que interesa, evitar el uso indebido de medicamentos, así como determinar la peligrosidad de éstos, su comprobada y comprobable acción y finalidades terapéuticas y sus ventajas científicas, técnicas o económicas, de acuerdo con los adelantos científicos (arts. 7, 8, 14 y concs. ley cit.; y 2, 13, 26, 27, 35 y concs. decreto cit.).
7. Que, a su turno, en uso de la facultad recordada (art. 40 decreto 9763/1964), el ministerio citado expidió la disposición 3916 (02/07/1985), tendiente a controlar la experimentación en el ser humano de productos farmacéuticos. Esta reglamentación admitía que los progresos registrados en los últimos años en la síntesis y elaboración de nuevos y potentes productos de uso farmacéutico hacía necesario un control adecuado tanto de su eficacia como de sus posibles efectos secundarios, máxime cuando, en general, la actividad farmacológica de aquéllos es acompañada por un incremento de sus efectos indeseables. De ahí que la administración de esas sustancias al ser humano deba ser precedida por una metodología de evaluación rigurosa, objetiva y segura, que la farmacología clínica brinda al paso que garantiza, una vez cumplidas sus distintas fases de investigación, que tales sustancias posean la actividad que se les atribuya, y seguridad a los sujetos en los que se ensayan. Es esta disposición, por lo demás, continuadora de otras dictadas con análogas finalidades (disposición 308 de la Subsecretaría de Medicina Social y Fiscalización Sanitaria, del 23/02/1983; resolución 858 de la Secretaría de Estado de Salud Pública, del 10/04/1979, entre otras).
Es esencial poner de relieve los numerosos requerimientos que deben satisfacer quienes pretenden realizar estudios e investigaciones de farmacología clínica.
Los proyectos que se presenten deberán demostrar fundamentalmente cuáles son las propiedades farmacológicas y terapéuticas del compuesto a estudiar relacionándolas en forma cualitativa y cuantitativa con el empleo terapéutico que se preconiza, así como cuál es el margen de sanidad y los efectos adversos previsibles en las condiciones de empleo terapéutico para el ser humano. También se exige, en su caso, la presentación de antecedentes bibliográficos, del plan experimental detallado y fundamentado -debiendo ser la metodología sensible al fin propuesto y exhaustivamente descripta- y la de los resultados obtenidos en su totalidad, consignándose gráficos, fotografías, tablas, cifras y todo elemento que permita su evaluación crítica independiente de la interpretación de los autores. Súmase a todo ello la exigencia de dar a conocer la discusión de los resultados obtenidos, debiendo interpretar los datos de manera que permita caracterizar el compuesto farmacológica y toxicológicamente poniendo de manifiesto sus acciones farmacológicas, sus efectos colaterales, el margen de seguridad que ofrece, etcétera. En suma, diversos y variados elementos que atañen a la "información general" y a la "información preclínica" que incluye la relativa a la farmacología animal - farmacodinamia, farmacocinética y toxicología animal -aguda, subaguda, crónica- para lo que se indican las pautas generales que deberán observarse acerca del tiempo de administración en los estudios toxicológicos.
8. Que, como se lo ha anticipado, la resolución n. 47 citada constituye un elemento relevante para esclarecer este debate, sobre todo cuando, por lo antedicho, es indiscutible que ha emanado del órgano al que la ley dejó el control de esta materia.
Es pertinente, entonces, analizar las razones en que este acto se apoyó para dar por finalizada la experimentación dispuesta por la resolución n. 522 citada, y que son las contenidas en sus considerandos, como expresamente indica su art. 1 . Surge de ello: "que la investigación, producción y suministro del compuesto, como la suspensión de su entrega, fueron ajenas a toda decisión del Ministerio de Salud y Acción Social. Que no obstante ello, este Departamento de Estado debe adoptar medidas vinculadas con el estado de necesidad generado por tales hechos. Que la circunstancia de hallarse comprometido en la investigación un número determinado de personas que presentan enfermedades carcinomatosas en estadio terminal, sujetas a la experiencia de su voluntad, exige la adopción de medidas extraordinarias para atender una situación bien atípica que no presenta otras vías para su abordaje. Que la investigación, por su naturaleza experimental, debe ser limitada, toda vez que la misma busca establecer los reales alcances farmacológicos de la crotoxina A y B y no fines terapéuticos. Que las disponibilidades de la sustancia son limitadas. . . Que tal bien jurídico 'que se intenta preservar' no es otro que el deseo supremo de preservar la vida humana. Que por todo lo señalado es necesario obtener una información evaluatoria de estricto carácter científico sobre la investigación, a efectos de determinar fehacientemente los reales alcances del compuesto aludido".
9. Que, en consecuencia, queda fuera de toda duda que la autoridad a la que ha sido otorgado el ejercicio del poder de policía sanitaria respecto de las actividades comprendidas en la norma de control de drogas y productos utilizados en medicina humana, y la facultad de dictar las disposiciones reglamentarias o complementarias que sean necesarias a tal fin, y que las ha ejercido respecto de la farmacología clínica, ha emitido su opinión sobre el complejo crotoxina A y B.
Asimismo, tal dictamen del órgano competente es inequívoco en cuanto a que, en las presentes circunstancias, ese complejo carece de acción antineoplásica.
Además ningún elemento de convicción obra en el expediente que pueda originar el excepcional supuesto que autorizaría a revisar la validez de la causa de ese acto administrativo.
10. Que de todo ello se sigue una consecuencia de la mayor importancia: no incumbe a los jueces en el ejercicio regular de sus atribuciones, sustituirse a los otros poderes del Estado en las funciones que les son propias, sobre todo cuando la misión más delicada de la justicia es la de saberse mantener dentro de la órbita de su jurisdicción, sin menoscabar las funciones que le corresponden a los otros poderes.
11. Que no es del caso señalar los fundamentos en que el principio se sustenta. Sí lo es, el de destacar un aspecto no lo suficientemente puntualizado.
La doctrina de la división de los poderes o la separación de las funciones, especialmente en nuestras sociedades modernas, halla también su causa y finalidad en la especialización que pide el cumplido ejercicio de las diversas funciones que deben satisfacer los estados. Luego, la distribución de dichas funciones en órganos, cuya integración personal y medios instrumentales está pensada con arreglo a la especificidad de aquéllas, es prenda de un mejor acierto de sus proyectos y realizaciones.
12. Que de todo ello ha de hacer mérito el Poder Judicial cuando es llamado a ejercer su ministerio. Del juicio prudente de los magistrados en torno de los alcances de su jurisdicción, es de donde cabe esperar los mejores frutos en orden al buen gobierno de la Nación.
13. Que corresponde, por tanto, señalar que está fuera de discusión que la actividad de la Administración en materia de drogas y productos medicinales así como su experimentación y suministro a los pacientes, lejos de menoscabar los derechos a la vida y a la salud, garantiza las condiciones más adecuadas y seguras para que tales derechos cundan.
Dicha actividad no sólo tiende a la preservación de esos valores, ante los eventuales efectos nocivos de alguno de los aludidos productos, sino que también se halla enderezada a evitar que el hombre pueda tornarse en el sufriente receptor de múltiples manipuleos sólo basados en la conjetura, la doxa o la improvisación; esto es, impedir que el sujeto se "cosifique" como objeto de una mera investigación.
14. Que, en tal sentido, la fiscalización estricta de la experimentación y subsiguiente comercialización de productos medicinales tiende a evitar que esa actividad científica y comercial derive en eventuales perjuicios para la salud.
15. Que cabe inferir, además, que el indelegable control que debe ejercer el Estado en este campo reconoce no sólo razones estrictamente científicas sino también el imperativo ético de no permitir la utilización del hombre como un simple medio para otros fines.
Es más, un adecuado paradigma de la salud no puede dejar de observar que, principalmente en enfermedades como el cáncer, sus efectos trascienden a la persona del paciente hasta conmover su núcleo familiar o de afectos, cuyos miembros, en muchas ocasiones resultan llamados a tener que prestar su opinión y consejo en trascendentes decisiones sobre el tratamiento a seguir. No es aventurado, entonces, afirmar que la protección del hombre enfermo a la que apuntan las normas citadas, debe entenderse que se extiende a todos los sujetos comprendidos en tales ámbitos.
16. Que una cuestión que guarda con la del sub lite un particular vínculo, fue resuelta por la Suprema Corte de Justicia de los Estados Unidos de América (United States v. Rutherford, 18/06/1979, 61 L. Ed. 2d. 68). El producto entonces en juego se denominaba "laetrile", proclamado como antineoplásico. Cabe observar que una de las diferencias que media entre ambos casos vuelve aún más interesante el precedente, toda vez que no se reclamaba en él el suministro y producción por el Estado de la droga, sino sólo que se prohibiese a éste, respecto de los enfermos terminales de cáncer, que impidiera el embarque interestatal y la venta de laetrile, una droga no aprobada para su distribución bajo la "Federal Food, Drug and Cosmetic Act", que vedaba la distribución entre los estados de toda "nueva droga", antes de que la Secretaría de Salud, Educación y Bienestar del Gobierno Federal aprobara su aplicación con fundamento en evidencias sustanciales sobre la seguridad y efectividad de aquélla.
Es de recordar, también, que la Cámara de Apelaciones había opinado que los términos "seguridad" y "efectividad" usados en ese Estatuto no tenían una razonable aplicación a los enfermos terminales de cáncer: desde que esos pacientes, por definición, "pueden morir de cáncer con indiferencia de lo que se les dé", no había estándares reales para medir la seguridad y efectividad de una droga para esa clase de individuos. Por ello, la Cámara confirmó el mandado (injunction) del tribunal de distrito, que permitía el uso de laetrile en enfermos de cáncer, cuyo carácter terminal se hallase comprobado.
La Corte norteamericana, al revocar ese pronunciamiento expresó: "que dentro de nuestro sistema constitucional, los tribunales federales no desempeñan la función de juntas revisoras con autoridad para rehacer las leyes según sus propias concepciones acerca de las directivas más adecuadas de política general. Sólo cuando la aplicación textual de la ley conduce a resultados tan irrazonables que no sería justo atribuirlos a la intención del Congreso, cabe que los jueces den por sobreentendida la excepción a la letra de la ley". Y, tanto en aquel caso como en éste, no se advierte que las normas que los rigen hayan dejado de proteger a los enfermos terminales de cáncer de las drogas no efectivas o inseguras.
Es del todo apropiado, no obstante su extensión, trascribir otros pasajes de la sentencia citada, dado que brindan elementos de gran valor ilustrativo. "Existe -continuó expresando la Corte norteamericana- un especial sentido en el cual la relación entre eficacia y seguridad de una droga tiene significación en el contexto de las enfermedades incurables. La inocuidad de una droga puede ser peligrosa para cualquier paciente si ella no produce los efectos terapéuticos implicados. . . Pero si un individuo que sufre de una potencial enfermedad fatal rechaza una terapia convencional en favor de una droga de propiedades curativas no demostradas, las consecuencias pueden ser irreversibles". En la nota a este párrafo, indica la Corte que, según la declaración del Dr. Carl Leventhal, director delegado de la oficina de fármacos, FDA, y profesor auxiliar de neurología y patología en la Universidad de Georgetown: "la seguridad de una droga para uso humano depende, en buena medida de la eficacia terapéutica de la droga de que se trata. En el caso del cáncer, el tratamiento con una droga no efectiva conducirá. . . necesariamente a la muerte del paciente"; y que, según la declaración del Dr. George J. Hill, presidente del departamento de cirugía y de la facultad de Medicina de la Universidad de Marshall, W. Va.: "un tratamiento ineficaz puede llevar a retardar el empleo de los medios terapéuticos reconocidos, provocando muertes innecesarias, por ello, en ausencia de evidencias científicas sobre su efectividad, ninguna droga destinada al tratamiento del cáncer puede ser considerada segura". A continuación, el tribunal agregó: "por esas razones, aun antes de que la enmienda de 1962 incorporase el estándar de eficacia a la tramitación requerida para la aprobación de nuevos medicamentos, la FDA consideraba la efectividad cuando revisaba la seguridad de las drogas usadas para tratar enfermedades terminales. La práctica de la FDA refleja, asimismo, el reconocimiento, ampliamente respaldado en este caso por los testimonios de médicos expertos, de que en enfermedades como el cáncer, muchas veces es imposible identificar a un paciente como terminal salvo retrospectivamente". En la nota a este pasaje se indican las declaraciones del Dr. Peter Wiernik, jefe de la rama de clínica oncológica del Instituto Nacional del Cáncer del Centro de Investigaciones de Baltimore: "Nadie puede prospectivamente definir la expresión 'terminal'con cierta exactitud. De un paciente se puede decir que es terminal sólo después de su muerte. Muchos pacientes en estado crítico responden a los modernos tratamientos de cáncer"; y la declaración del Dr. Joseph Ross, profesor de medicina, de la Escuela de Medicina de la Universidad de California, en Los Angeles: "La distinción entre pacientes 'terminales'y 'no terminales'no puede ser establecida con precisión y presuponer que laetrile puede ser suministrado a pacientes terminales con impunidad podría privar a esos pacientes de otros medios terapéuticos que pueden ayudarlos". "El cáncer en sus distintas formas -añadió la Corte norteamericana- varía considerablemente en su comportamiento y en su respuesta a las diferentes terapias", y recordó en la nota pertinente: "El comisionado advirtió que estos cuadros de comportamientos imprevistos pueden explicar las alegaciones de base anecdótica acerca de la efectividad del laetrile. Personas tratadas con laetrile que experimentaron una espontánea mejoría o que respondieron tardíamente a las terapias convencionales después de haberlas abandonado, pueden creer que su mejoría se debe al laetrile. . . Sobre todo desde que tratamientos del cáncer probados como la quimioterapia y la radiación tienen a menudo efectos dolorosos, el comisionado concluye que los pacientes que subjetivamente perciban un progreso después de sustituir por laetrile a esos tipos de terapia pueden erróneamente creer que su condición se ha vuelto estacionaria o que ha mejorado". Y sigue la Corte: "Incluso individuos en estado crítico pueden experimentar una inesperada reacción y comenzar a responder al tratamiento acostumbrado. Por ello, como lo concluye el comisionado, permitir esta excepción a lo dispuesto por la ley, sin que medie prueba de la eficacia del producto en el tratamiento del cáncer 'puede provocar muertes inútiles y el sufrimiento en los pacientes caracterizados como terminales que en la actualidad podrían ser auxiliados por medio de terapias comprobadas'. Aceptar la proposición de que los estándares de seguridad y eficacia del 'act'no tienen relevancia para los pacientes terminales es negar la autoridad del comisionado sobre todo medicamento que se suministre a esas personas, por más tóxico o ineficaz que sea. Si la historia suministra alguna guía, este nuevo mercado no debe ser tolerado. Desde el comienzo de este siglo, afanosos fabricantes y comerciantes han anunciado una extensa variedad de, presuntamente, sencillas e indoloras curas para el cáncer, incluyendo linimentos de terpentina, mostaza, aceite, huevo y amoníaco; musgo de pantano; combinaciones de reflectores de colores; pastas hechas con glicerina o queso de Limburgo; tabletas minerales, y mezcla de la 'Fuente de la juventud', hecha con especias, aceite y grasa. Al citar estos ejemplos, no tenemos, por supuesto, la intención de menoscabar la sinceridad de los que en la actualidad proponen la utilización de laetrile, ni ello involucra ningún juicio acerca de si dicha droga puede finalmente resultar un medio seguro y efectivo en el tratamiento del cáncer. Precisamente. la experiencia de la historia indica por qué el Congreso pudo razonablemente proponerse proteger a los enfermos terminales, en no menor medida que a otros pacientes, del amplio espectro de autoproclamadas panaceas que el ingenio humano puede elaborar".
17. Que, en conclusión, corresponde decidir que en el sub exámine no se encuentra observado uno de los recaudos que, supuesto el derecho subjetivo constitucional de que se trata, condicionaría inexcusablemente la invocación legítima de éste (consid. 4).
Desde luego, ello no implica negar las eventuales propiedades antineoplásicas del complejo crotoxina A y B que puedan ser demostradas científicamente en el futuro. El juicio asertado sólo expresa que, según los motivos puestos de manifiesto por el órgano legalmente autorizado, dicha sustancia no provoca, aquí y ahora, tales efectos y que, salvo que se acredite inequívocamente la inexactitud de esos fundamentos, los jueces deben atenerse a ellos.
Por ello, de conformidad en lo pertinente con lo dictaminado por el procurador general, se hace lugar al recurso extraordinario y se confirma la sentencia apelada.- Augusto C. Belluscio.- Enrique S. Petracchi.- Jorge A. Bacqué.
PAGINA 12 - 22 DE ABRIL DE 2007
LA JUSTICIA FRENO UNA LICITACION Y PROHIBIO HASTA PRODUCIR UN GENERICO
Lo que importa acá es el negocio
Un juez aceptó una demanda del laboratorio multinacional Squibb y prohibió que un argentino fabricara siquiera didanosina, un antirretroviral para pacientes con VIH, por cuestiones de patentes. El Gobierno lo considera un “abuso” y una amenaza al sistema de salud. El laboratorio ya fue punido en EE.UU. por prácticas monopólicas similares.
Final del formulario
El conflicto es considerado un caso testigo en el país. El juez Alejandro Saint Genez, del juzgado Nacional Civil y Comercial Nº 9, dictó una medida cautelar en favor de un laboratorio farmacéutico extranjero. El fallo prohíbe a los laboratorios nacionales producir y comercializar un medicamento para pacientes con VIH Sida. La resolución tiene que ver con una cuestión de patentes. Afecta la fabricación del antirretroviral didanosina, una droga que en la Argentina toma el 6 por ciento de los infectados con el virus (unos 1800 pacientes). El caso, que fue denunciado por los laboratorios nacionales y el Gobierno como un “abuso” que amenaza el sistema público de salud, abre el debate sobre las prácticas monopólicas de las multinacionales de medicamentos con respecto a las drogas de las que depende la vida o la muerte de las personas: aunque esta discusión concreta es por un remedio para los enfermos de sida, está sucediendo lo mismo con los medicamentos oncológicos. El efecto de las prácticas denunciadas es la suba artificial de los precios de los medicamentos.
El ministro de Salud, Ginés González García, dio un dato para dimensionar los intereses económicos en juego: señaló que la decisión del juez Genez sobre la didanosina obligará al Estado a gastar “2,5 millones de pesos más por año por la misma droga” que ya se fabrica localmente.
El funcionario consideró que la patente en base la cual se dictó la cautelar es dudosa. “Lo que está pasando es que el sistema de patentes para muchas drogas se está agotando, porque con los años caducan. Entonces algunos laboratorios, como sucedió en este caso, realizan una pequeña innovación para decir que tienen una droga nueva y conseguir así una nueva patente. Son maniobras que les permiten mantener artificialmente sus derechos exclusivos por varios años más y fijar altos precios”, dijo el ministro a Página/12.
González añadió que existe “una estrategia” de las multinacionales farmacéuticas que “en una maniobra con estudios jurídicos” buscan de esta manera “esquivar la ley” y asegurarse su posición monopólica en el mercado. Y calificó el fallo como una “extorsión a la democracia”.
La controversia tiene origen en una licitación de diciembre pasado, cuando el ministerio llamó a un concurso para adquirir una partida de didanosina, uno de los 17 antirretrovirales que el Estado provee a los pacientes que carecen de cobertura social. A la licitación se presentó un único oferente, el laboratorio Richmond, de capitales nacionales. Pero su presentación fue impugnada por Bristol Myers Squibb, dueño de la patente de la didanosina. A mediados de enero, haciendo lugar a tal solicitud, el juez Saint Genez dictó la medida cautelar prohibiendo la fabricación local de la droga y su venta, con lo que dejó desierta la licitación.
Esta semana, los laboratorios nacionales agrupados en la Cámara Industrial de Laboratorios Farmacéuticos Argentinos (Cilfa) y la Cámara Empresaria de Laboratorios Farmacéuticos (Cooperala) dieron al conflicto difusión pública, con una solicitada en la que condenaron el fallo judicial que, dijeron, “genera monopolios y pone en riesgo la provisión de medicamentos”.
En cambio, la Cámara Argentina de Especialidades Medicinales (Caeme), a la que pertenece Bristol, defendió la decisión del juez: “Las patentes no generan monopolios; por el contrario, alientan la competencia por generar productos innovadores en beneficio de la sociedad”, señaló el organismo en una gacetilla distribuida a la prensa.
Derechos
Carlos Correa es profesor de la UBA, donde dirige el posgrado en Propiedad Intelectual de la Facultad de Derecho. El especialista explicó que la didanosina “no fue desarrollada por el laboratorio Bristol, sino por el Instituto del Cáncer de los Estados Unidos, que le otorgó a Bristol una licencia exclusiva para su comercialización. En el exterior, las patentes del producto ya han vencido. Lo que hicieron en la Argentina fue conseguir una patente registrando no la droga básica sino una forma particular que le dieron a la tableta de didanosina. Esa pequeña modificación les alcanzó para obtener derechos, con los que impiden la competencia de mercado y anulan la licitación del ministerio”.
Para Correa, “el fallo es un antecedente muy negativo por el tipo de producto de que se trata y por el hecho de que el juez otorgó la medida cautelar sin seguir el procedimiento que establece la ley”.
“El juez omitió un paso esencial: no llamó a un perito de oficio para dictar su fallo. Es decir que otorgó la cautelar en base solamente a lo que dijo el perito del laboratorio extranjero. Estamos ante el caso de una patente cuya validez es dudosa, un uso que va más allá de lo que sería permisible.”
Antecedentes
El laboratorio Bristol Myers Squibb tiene antecedentes de abuso del sistema de patentes en los Estados Unidos. En marzo del 2003, la Comisión Federal de Comercio (FTC) del gobierno norteamericano -equivalente a la Secretaría de Comercio nacional- comunicó que el laboratorio tuvo que aceptar un arreglo extrajudicial millonario por prácticas dudosas. Lo que hizo Squibb fue obstruir durante los años `90 la venta de genéricos que competían con tres productos de gran venta del laboratorio: dos drogas anticancerígenas, el Taxol y el Platinol, y el ansiolítico BuSpar.
De acuerdo a la comisión, Squibb hizo esto para quedarse con un mercado de casi dos mil millones de dólares anuales de estas drogas, que son muy caras, con lo cual forzó a los pacientes a pagar sobreprecios de cientos de millones de dólares.
La información está publicada en la página de la FTC, donde se lee que la investigación puso al descubierto que Bristol, entre otros trucos, le pagó 70 millones de dólares a otro laboratorio para que no pusiera en el mercado su genérico.
Las patentes
El otorgamiento de una patente tiene tres requisitos: que se trate de una innovación, que tenga altura inventiva y que sea de aplicación industrial. Desde Cilfa manifestaron que la sentencia del juez Genez está apelada. Eduardo Franciosi, director ejecutivo de la Cámara, explicó que el eje del recurso judicial es “el reclamo de que no se concedan patentes sin altura inventiva y que eso no sea utilizado luego ante la Justicia para constituir monopolios”.
El directivo señaló que Cilfa “respeta la ley de patentes argentina”, aunque quienes no pertenecen al mundo de los laboratorios medicinales podrían fácilmente encontrar más de una objeción a este marco legal.
Hasta la década pasada, la ley argentina no reconocía patentes a los medicamentos, de manera que era posible que distintos laboratorios los fabricaran y hubiera competencia. Esto cambió drásticamente cuando el país adhirió a los compromisos surgidos de la Organización Mundial de Comercio. “El acuerdo internacional de propiedad intelectual de la OMC no permite excluir de la patente una serie de productos”, más allá de cuál sea el valor humanitario de su aplicación, explicó Correa. Se trata de condiciones “negociadas en el ámbito del GATT, entre los años 1986 y 1994, e impulsadas por los países desarrollados, con una fuerte presión de la industria farmacéutica y del software”.
Clara Suárez, de Cooperala, hizo una lectura propia de las consecuencias. “La ley de patentes y la OMC aseguran que los derechos exclusivos sirven para apuntalar el desarrollo de la investigación y el mejoramiento tecnológico. A mi criterio, produce todo lo contrario, trabar el desarrollo, porque el conocimiento queda oculto y en manos de unos pocos.”
Los defensores del sistema de patentes sostienen, contra este argumento, que si no existen límites las empresas que no invierten en investigación se apropiarían del conocimiento generado por las que sí lo hacen. Como sea, esa discusión excede la situación abierta con el fallo sobre la didanosina, donde lo que está en cuestión es la validez de la patente que otorgó el propio Estado al laboratorio extranjero, y el posterior uso de esa ventaja en una maniobra sospechada de ser el primer paso de una ofensiva mayor.
CONCLUSIÓN
El sector de medicamentos reviste una importancia central en el país. Por un lado, como actividad económica involucra un mercado que en el 2004 registró un giro del orden de los 9.765 millones de pesos o 3,1% del PIB. Genera y sustenta miles de puestos de rabajo productivos y a la vez es intensivo en el uso de tecnología. En otras palabras se trata del tipo de sectores de actividad que suelen ser protegidos e incentivados en los países que buscan consolidar su industrialización.
Por otro lado, la utilización de medicamentos por parte de la población constituye el cuidado de salud más frecuente. Las encuestas de utilización y gasto en salud que desarrolla el Ministerio de Salud así lo prueban. Se producen 30,3 millones de actos de compra de medicamentos en farmacia por mes. Casi el 70% declara haber utilizado medicamentos durante el últimos mes, contra un 40% que dice haber realizado consultas médicas y un 5% que concurrió a un psicólogo. Incluso unos 3,2 millones de argentinos que declaran no percibir ingresos afirman consumir medicamentos.
Pero el problema reside en que “nadie puede servir a dos amos”. Es muy difícil conciliar la lógica que impera para un bien industrial de consumo masivo y la lógica necesaria para la adecuada producción y consumo de un bien social que genera salud. Es por eso que hay muchos mitos y confusiones en relación a los medicamentos, su producción, circulación y utilización.
El primero de estos mitos afirma que se trata de un mercado opaco y muy imperfecto. Bueno, en realidad todos los mercados son imperfectos ya que presentan “fallas”. La competencia perfecta es una imagen simbólica, un ideal que no se concreta en ningún caso. (Los mercados no fallan, quienes fallamos somos nosotros al creer que ellos resuelven de forma equitativa nuestras necesidades de salud). Pero si comparamos al mercado de los medicamentos con otros mercados de salud en Argentina, verificamos que es bastante más competitivo y transparente que el resto. Por ejemplo, se identifican como fallas que existen diferentes precios para el mismo producto (dispersión), que la formación de precios es arbitraria y hay asimetrías de información en el momento de definir la compra. Estas fallas son mucho mayores en otros mercados como el de la consulta ambulatoria, los tratamientos psicológicos, las internaciones y la tecnología médica. En la mayoría de los casos en estos items ni siquiera puede el comprador conocer el precio antes de realizar la adquisición. No existen listas de precios que permitan comparaciones y el mismo proveedor, cobra valores diferentes a diferentes compradores, por el mismo bien o servicio.
Otro mito afirma que el gasto en medicamentos en el país es muy alto. Los estudios demuestran que el gasto de medicamentos de un país es relativo al tamaño de la economía, a la evolución demográfica y epidemiológica, a los esquemas de financiamiento en salud y, sin duda, a los precios medios vigentes. Cuando se considera todas estas variables se concluye que Argentina no tiene un nivel de gastos sobredimensionado en medicamentos.
Es que “el problema no se ven en la foto sino en la película”. El ritmo con que crece el gasto en medicamentos supera al del gasto en salud. Las curvas de evolución de ambos gastos son diferentes y este es un fenómeno que se da tanto en Argentina como en otros países.
También vinculado con el gasto se debe destacar que más que un problema por el nivel excesivo del mismo tenemos un problema por la composición de su financiamiento. En la actualidad, alrededor del 70% del gasto en medicamentos en el país se financia con recursos de las familias (gasto de bolsillo) y esta es la forma más regresiva e injusta de financiarlo.
Es innegable que la situación económico-social de fines del año 2001 y principios de 2002, necesitaba una acción inmediata por parte del estado en ejercicio de su poder de policía.
Pues bien, tal reacción no se hizo esperar. El Programa Remediar es una intento de facilitar el acceso a medicamentos a los sectores que no podían afrontar los costos de los fármacos.
Si hay algo que caracteriza históricamente el obrar del Estado en este tipo de circunstancias es un triste trípode: 1) alto nivel de burocracia; 2) falta de transparencia en el manejo de fondos; y 3) limitado acceso que frustra el objetivo.
Más allá de las críticas enunciadas todo hace parecer que este plan no es un ejemplo más de un Estado ineficiente y corrupto.
Si bien la entrega de medicamentos tiene un cierto nivel de complicación, éste no llega a ser tal que el supuesto beneficiario termine “mareado” por las tramitaciones. Es decir, si bien hay tramites necesarios, éstos no implican un nivel de burocratización que atente contra la eficiencia del plan.
En lo referente a la falta de transparencia, este punto es el más flojo del proceso desde los sitios oficiales no se brinda información clara y es justamente en este ítem donde se concentran las críticas más y mejor fundadas.
El impacto social no recibe críticas de entidad suficiente como para opacar los logros conseguidos. Aproximadamente 13 millones de personas que acceden gratuitamente a medicamentos de suma importancia es un dato no menor. Sería interesante que el vademécum sea ampliado y se tomen en cuenta políticas de salud reproductiva, las cuales son un tema siempre vigente.
En resumen podría decirse que el concepto del plan es positivo en líneas generales.
Punto 1): aprobado con salvedades
Punto 2): a recuperar
Punto 3): aprobado con salvedades
La única triste conclusión a la que arribamos es que nuevamente el centro de la crítica recae en el manejo desprolijo o poco claro de los fondos empleados.
En cuanto al patentamiento, en el campo farmacéutico presenta problemas complejos, no sólo por su intensidad y la variedad de modalidades que asume sino por sus implicaciones sobre el acceso a medicamentos, especialmente por parte de quienes menos recursos tienen. Los países en desarrollo tienen, dentro de lo prescripto por los tratados internacionales, la posibilidad de establecer los estándares de patentabilidad en un modo que se asegure la protección de las invenciones genuinas, impidiendo al mismo tiempo la indebida limitación de la competencia legítima.
BIBLIOGRAFÍA
Artículos periodísticos
Páginas web
Administración Nacional de Medicamentos, Alimentos y Tecnología Médica
Ministerio de Salud de la Nación Argentina
Programa Nacional Remediar
Editorial Lexis Nexis
Pagina oficial de Normas
Pagina web del Dr. Federico Tobar
Superintendencia de Servicios de Salud
Presidencia de la Nación Argentina
Biblioteca Virtual de Salud
Banco Interamericano de Desarrollo
Dr. Pedro M. Politi; “Plan Remediar”
101
Fuente: Cátedra Libre de Salud y Derechos Humanos de la Facultad de Medicina -con datos del Indec-
Fuente: Elaboración propia basado en un gráfico publicado en www.dolarsi.com.ar -elaborado con datos del www.econline.com.ar-
Fuente: www.remediar.gov.ar
Descargar
| Enviado por: | El remitente no desea revelar su nombre |
| Idioma: | castellano |
| País: | Argentina |
Todos los derechos reservados.