Salud
Fibrosis quística
Introducción
Clásicamente la fibrosis quística se ha considerado una enfermedad propia de la infancia. En estos últimos años ha perdido esta característica por lo que, actualmente, debe ser una enfermedad bien conocida por los internistas, neumólogos y gastroenterólogos. La razón de este cambio ha sido doble: en primer lugar, la mayor expectativa de vida de estos pacientes debido a las mejoras terapéuticas introducidas, en particular un mejor tratamiento de las infecciones respiratorias y del estado de nutrición, y, en segundo lugar, porque algunos pacientes, debido probablemente a una menor penetración de su alteración genética, presentan unas manifestaciones clínicas más solapadas, por lo que son diagnosticados en la pubertad o incluso en la edad adulta.
Etiopatogenia
La fibrosis quística es la enfermedad hereditaria más común de la raza blanca. Se transmite con carácter autosómico recesivo y afecta a uno de cada 2.000 nacidos vivos de raza blanca y a uno cada 17.000 nacidos vivos de raza negra. Los pacientes tienen un riesgo de engendrar un hijo enfermo de uno de cada 100, cifra muy superior a la de la población general. Existe aproximadamente un 5% de portadores sanos. El gen responsable de la enfermedad fue clonado en 1989. Se localiza en la región 31 del brazo largo del cromosoma 7 y codifica una proteína de 1.480 aminoácidos denominada reguladora de la conductancia transmembrana de la fibrosis quística (CFTR). La mutación más común es de tres bases en el codón 508, que da lugar a la pérdida de una fenilalanina (DF508), aunque existen alrededor de 150 mutaciones más. La mutación DF508 afecta al 80% de las fibrosis quísticas de Norteamérica y del norte de Europa y al 50% de las mediterráneas. Así pues, las restantes mutaciones son mucho menos frecuentes. La CFTR es una glucoproteína que regula el transporte del cloro a través de 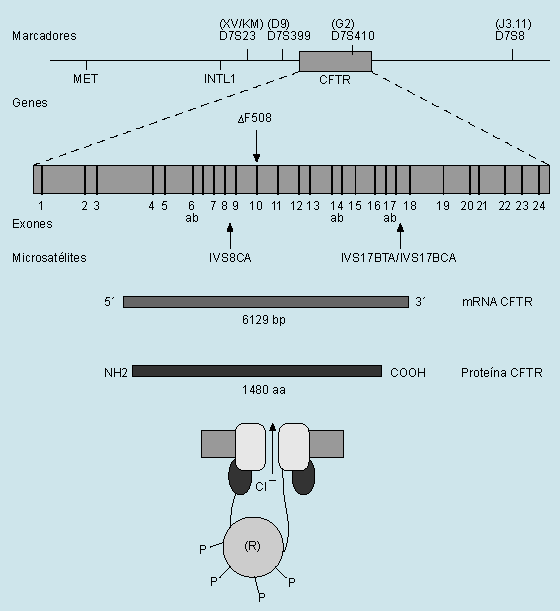
la membrana de las células epiteliales exocrinas. El paso del cloro ocurre por unos canales que se abren en respuesta al incremento de los niveles celulares de AMPc, el cual, a su vez, activa la enzima proteincinasa. Esta sustancia facilita el paso del cloro mediante la fosforilación de la proteína CFTR. Cuando ésta se halla alterada, se produce un trastorno de la regulación de los canales del cloro, que se traduce en una disminución de la secreción de agua, manteniéndose la misma cantidad de proteína. Esto conduce a un aumento de la viscosidad de las secreciones y a una disminución de la motilidad del órgano afectado. Como consecuencia se produce una obstrucción de los conductos de los órganos que poseen células epiteliales exocrinas, como el pulmón, el páncreas, el intestino y las glándulas sexuales.
En las glándulas sudoríparas de la fibrosis quística la conductancia del epitelio de los conductos se halla reducida a un nivel casi indetectable y, como consecuencia, los iones sodio y cloro son pobremente reabsorbidos, lo que origina un elevado contenido de sal en el sudor. Esta alteración es la que permite establecer el diagnóstico de la enfermedad.
Cuadro clínico
La fibrosis quística es una enfermedad multisistémica en la que se hallan involucrados los aparatos digestivo, respiratorio y reproductor.
Aparato digestivo
Esta enfermedad afecta potencialmente todos los órganos abdominales con función secretora. La función exocrina del páncreas y del intestino están constantemente comprometidas, mientras que el hígado suele estarlo con menor frecuencia. La afección intestinal se puede manifestar en el mismo momento del nacimiento en forma de íleo meconial. Éste se presenta en el 10% de las fibrosis quísticas y se manifiesta en forma de obstrucción intestinal provocada por el aumento de viscosidad de las secreciones intestinales. Ello se debe, por un lado, a la existencia de una escasa secreción de cloro de las criptas intestinales, con una reabsorción vellositaria normal de este ion; así, se ve alterado el transporte del agua, lo que provoca una disminución de la fluidez del contenido intestinal. Por otro lado, existe una falta de secreción de las enzimas pancreáticas que en condiciones normales digieren y fluidifican el meconio.
El cuadro clínico se caracteriza por dolor y distensión abdominal, vómitos y ausencia de deposiciones meconiales durante los primeros días del nacimiento. Se puede asociar a atresia del intestino delgado o a vólvulo de íleon. La mortalidad, que años atrás era del 55%, ha descendido al 5% en los últimos 20 años, gracias a la introducción de modernas técnicas quirúrgicas. En niños mayores y en adultos puede producirse un prolapso rectal a causa de la dificultad de evacuación y, en casos extremos, una obstrucción intestinal por impactación fecal, debido a un mecanismo similar al del íleo meconial.
La afección pancreática se presenta en el 90% de los enfermos y se traduce clínicamente por insuficiencia pancreática exocrina, cuya consecuencia final es la esteatorrea. Esta alteración se presenta porque existe una disminución de la excreción de agua y bicarbonato, lo que determina un aumento de viscosidad del jugo pancreático. Como consecuencia de ello se producen obstrucciones ductales, con formación de quistes y, por último, atrofia glandular, infiltración grasa de la estroma y fibrosis. La malabsorción de grasas es potenciada por un aumento de las pérdidas de sales biliares por las heces, lo que dificulta la formación de la fase micelar. La absorción de los hidratos de carbono está poco afectada, dado que el déficit de amilasa pancreática es parcialmente compensado por la secreción de amilasa salival. La insuficiencia pancreática es responsable de intensa malnutrición y se presenta en las fases iniciales de la enfermedad, de forma más acusada en los pacientes diagnosticados más precozmente, habiéndose observado más relacionada con la mutación DF508.
El hígado y las vías biliares también se ven involucrados en esta enfermedad, debido al trastorno de la secreción en los conductillos biliares, que llegan a dilatarse; luego se desarrolla fibrosis y, finalmente, puede instaurarse una cirrosis biliar con signos de hipertensión portal, aunque esta afección sólo la padece el 1% de los pacientes con fibrosis quísticas. Por esta razón se recomienda practicar una prueba del sudor a todo paciente con cirrosis biliar de causa no aclarada. Debe tenerse en cuenta que el 30% de los pacientes mayores de 10 años presentan hepatomegalia, y el 10%, alteraciones de la función hepática. En los adultos se ha observado una alta frecuencia de litiasis biliar debida a la pérdida intestinal de sales biliares.
Aparato respiratorio
Las manifestaciones respiratorias se deben al aumento de la viscosidad de las secreciones bronquiales y a un aclaramiento mucociliar defectuoso, que provocan una impactación mucosa e infección crónica de las vías aéreas periféricas, lo que condiciona el desarrollo de bronquiectasias, fibrosis peribronquial y, finalmente, obstrucción bronquial debido a la presencia de un proceso inflamatorio crónico con liberación de enzimas proteolíticas. La lesión pulmonar evoluciona hacia una insuficiencia respiratoria con importante hipoxemia y, a la larga, al cor pulmonale.
Los gérmenes responsables de las recidivas de las infecciones son Pseudomonas aeruginosa, Staphylococcus aureus y, con menor frecuencia, Haemophilus influenzae, estreptococo, Klebsiella, Chlamydia y Mycoplasma. Aunque el grado de insuficiencia respiratoria se ha observado ligada a la mutación DF508, y de forma especial a los pacientes homocigotos, el mayor tiempo de evolución de la enfermedad es otro factor determinante, debido a que estos pacientes han sufrido infecciones respiratorias de forma más reiterada. Otras posibles alteraciones del aparato respiratorio son hemoptisis (5%), neumotórax, rinosinusitis y poliposis nasal.
Aparato reproductor
Los pacientes varones presentan azoospermia por aplasia de las vesículas seminales, del epidídimo y del cordón espermático, de forma que muchos de ellos son estériles. Las mujeres, a pesar de presentar un aumento de la viscosidad del moco cervical, son más fértiles que los hombres.
Diagnóstico
En este apartado debe considerarse el diagnóstico clínico, ante la presencia de los síntomas descritos, los diagnósticos neonatal y el prenatal.
Generalmente las manifestaciones clínicas que primero llaman la atención son las respiratorias, aunque la esteatorrea, si es muy intensa, o un retraso acusado del desarrollo también pueden hacer sospechar el diagnóstico. Para confirmar la enfermedad se realizará la prueba del sudor, que consiste en provocar un exceso de sudación mediante una inyección de pilocarpina en una zona de la piel, donde se determina la concentración de cloro y sodio. Para confirmar el diagnóstico en los niños la concentración de cada uno debe ser superior a 60 mEq/L, pero en los adultos debe superar los 90 mEq/L. Aproximadamente el 98% de los pacientes dan positivo a esta prueba.
Debido a que el tratamiento precoz de la sintomatología mejora el pronóstico de la enfermedad, es conveniente realizar el diagnóstico precoz en el período neonatal en aquellos casos en que se sospecha la enfermedad o existan antecedentes familiares.
En la actualidad se utiliza para ello la determinación de la tripsina sérica por radioinmunoanálisis, que presenta valores anormalmente elevados dentro del primer año de vida, para ir descendiendo, en fases posteriores, a niveles inferiores a la normalidad, en relación con el grado de deterioro de la función exocrina pancreática. El diagnóstico prenatal puede realizarse mediante la determinación de la actividad gammaglutamiltranspeptidasa y de la fosfatasa alcalina en el líquido amniótico de la madre, aunque desde hace un tiempo es posible hacerlo mediante amplificación del DNA y electroforesis en geles de poliacrilamina.
Diagnóstico diferencial
El diagnóstico diferencial debe hacerse en el caso de íleo meconial con la atresia intestinal, la enfermedad de Hirschsprung, la invaginación intestinal, la atresia congénita de los conductos pancreáticos y el síndrome de Shwachman. Cuando el paciente presenta diarreas y malabsorción, es necesario efectuar el diagnóstico diferencial con la enfermedad celíaca, la deficiencia de disacaridasas, las enteropatías perdedoras de proteínas y el déficit de tripsinógeno o enterocinasa.
En caso de predominar la sintomatología respiratoria debe descartarse el asma, las bronquiectasias, la fístula traqueoesofágica y las alteraciones anatómicas vasculares.
Tratamiento
El tratamiento va dirigido a combatir las complicaciones de la enfermedad y a mejorar la calidad de vida del paciente, ya que, dada su etiología, no se dispone de un tratamiento curativo. Malnutrición y problemas del aparato digestivo. La malabsorción de grasas y las infecciones respiratorias de repetición conducen a los pacientes con fibrosis quística a un importante estado de malnutrición. Éste se valorará a través de determinaciones seriadas de datos antropométricos (peso, talla, perímetro braquial y grosor del pliegue) y bioquímicos (hemograma, proteínas, albúmina, lípidos, tasa de protrombina).
La dieta será hipercalórica, rica en hidratos de carbono y proteínas, con un contenido normal de grasas. Si existe esteatorrea se añadirán preparados comerciales de enzimas pancreáticas en forma de microsferas con cobertura entérica, con el fin de evitar su desnaturalización por el ácido gástrico. La dosis aconsejada de lipasa es de 30.000-40.000 U en cada comida (3-4 cápsulas por ingesta), aunque debe adaptarse a las necesidades de cada individuo comprobando la reducción de la esteatorrea con el tratamiento. En caso de no conseguirse este resultado se reducirá el contenido grasode la dieta y se añadirán triglicéridos de cadena media y suplementos de vitaminas liposolubles.
El tratamiento del íleo meconial consiste en la cirugía, administrándose durante la intervención y en el postoperatorio inmediato un agente mucolítico, la N-acetilcisteína, en solución al 10%, a dosis de 5-10 mL, 3 veces al día por vía oral o en forma de enemas, para reducir la viscosidad del contenido intestinal. Aparato respiratorio. La finalidad del tratamiento de los problemas respiratorios es fluidificar las secreciones y prevenir y tratar las infecciones. La fluidificación de las secreciones pretende evitar la obstrucción bronquial, para lo cual se aconseja la fisioterapia respiratoria con medidas posturales que faciliten el drenaje (2-3 sesiones diarias de 20 min, antes de las comidas), la administración intermitente de broncodilatadores en aerosol y mucolíticos. Para tratar la infección respiratoria se administrarán antibióticos, que se elegirán según el antibiograma del esputo y se mantendrán hasta una semana después de que el enfermo haya alcanzado una función pulmonar semejante a la que tenía antes de la infección. Si las infecciones son persistentes, el tratamiento antibiótico debe ser continuado, pero si son poco frecuentes no está justificado un tratamiento profiláctico. En pacientes muy evolucionados con insuficiencia respiratoria grave puede indicarse el trasplante pulmonar, tras una prudente selección del candidato. La supervivencia en estos casos es del 60% a los 3 años, según los resultados analizados hasta 1991 en 200 pacientes trasplantados.
En la actualidad se está investigando el uso del diurético amilorida con el fin de inhibir la reabsorción del ion sodio de las vías respiratorias, por un mecanismo similar al del epitelio renal. Finalmente, se está ensayando el empleo de terapia genética utilizando vectores de adenovirus recombinantes para modificar el epitelio bronquial de estos pacientes. Dado el carácter crónico de la enfermedad, se aconseja procurar una asistencia psicológica, tanto para el paciente como para la familia.
Pronóstico
La expectativa de vida de estos pacientes ha mejorado sensiblemente en estos últimos años, pues ha pasado de 4 años en 1950 a 25 años en 1990. Ello se ha debido a un diagnóstico más precoz, a las mejoras en el soporte del estado de nutrición y a los avances en el tratamiento de las infecciones respiratorias. A pesar de ello, la causa más frecuente de muerte suele ser la malnutrición provocada por la maldigestión secundaria a la insuficiencia pancreática exocrina, la reiteración de las infecciones respiratorias y la falta de apetito.
Otras causas de muerte menos frecuentes son el íleo meconial, la insuficiencia respiratoria crónica, el cor pulmonale y la hepatopatía crónica. El pronóstico está claramente relacionado con la presencia de la mutación genética DF508, especialmente en los individuos homocigotos, ya que se asocia a una aparición más precoz de la sintomatología y una mayor alteración de la función pancreática y respiratoria.
Bibliografía
-
DI SANT'AGNESE PA, DAVIS PB. La fibrosis quística en el adulto. Am J Med 1979; 66: 121-132
-
FIEL SB. Manejo clínico de la patología pulmonar en la fibrosis quística. Lancet 1993; 341: 1.070-1.074
-
TIZZANO EF, BUCHWALD M. Fibrosis quística: más allá de la terapia génica. J Pediatr 1992; 120: 337-349.
-
NAVARRO COLÁS S., FARRÉ VILADRICH A., Fibrosis quística, en: FARRERAS - ROZMAN, Medicina Interna, Vol. I, 1.998
ANOMALÍAS DEL DESARROLLO FIBROSIS QUÍSTICA
Página 10
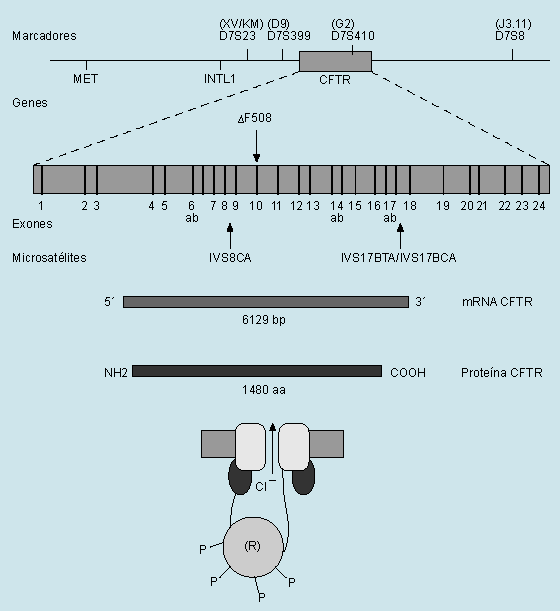
Fig. 1. Mapa del gen de la fibrosis quística con representación de sus exones (barras verticales), la mutación principal (DF508), los marcadores polimórficos, microsatélites y otros clones clave en el análisis del gen CFTR (cystic fibrosis transmembrane conductance regulator gene), el mRNA (trazado sombreado claro), la proteína CFTR (trazado sombreado oscuro) y representación de la estructura CFTR con los distintos dominios (transmembránico), unión al ATP y regulador (con unión de grupos fosfato, P).
Descargar
| Enviado por: | Luis Ignacio García Ventura |
| Idioma: | castellano |
| País: | España |
Todos los derechos reservados.