Química
Espectroscopia de absorción atómica
ESPECTROSCOPIA DE ABSORCIÓN ATÓMICA.
La espectroscopia de adsorción atómica usa la adsorción de la luz para medir la concentración de la fase gaseosa de átomos. Ya que la mayoría de las muestras son sólidas o líquidas, los átomos o iones de los analitos deben ser vaporizados a la flama o en un horno de grafito. Los átomos adsorben luz visible o ultravioleta y hacen transiciones a niveles de energía más altos. La concentración del analito es determinada por la cantidad de adsorción. Aplicando la ley de Beer-Lambert directamente en la espectroscopia AA es difícil debido a la eficiencia de la atomización de la muestra de la matriz y a la no-uniformidad de la concentración, y a la longitud de la trayectoria de los átomos del analito (en el horno de grafito AA). Las mediciones de concentración son generalmente determinadas de una curva de calibración, después de haber calibrado el aparato con los estándares de concentración conocida.

Instrumentación
Fuente de luz.
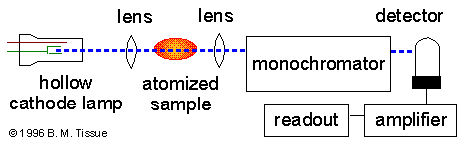
La fuente de luz usualmente es una lámpara de cátodo con vacío de los elementos a ser medidos. Los láseres son también usados en estos instrumentos. Los láseres son suficientemente intensos para excitar los átomos a mayores niveles de energía, esto permite a las mediciones de AA y fluorescencia atómica en un solo instrumento. La desventaja de estas angostas bandas de luz es que solo se puede medir un elemento a la vez. Los espectrómetros de AA usan monocromadores y detectores de luz visible y UV. El principal propósito de un monocromador es separar la línea de absorción del fondo de la luz debido a las interferencias. Los instrumentos de AA reemplazan a los monocromadores con filtro de interferencia band pass. Los tubos fotomultiplicador son comúnmente usados como detectores de espectroscopia AA.
Atomizador.
La espestroscopía de AA necesita que los átomos se encuentren en fase gaseosa. Los átomos y iones de la muestra deben sufrir desolvación y vaporización a altas temperaturas como en el horno de grafito o la flama. La flama de AA solo puede ionizar soluciones analíticas, mientras que el horno de granito puede aceptar soluciones, mezclas o muestras sólidas.
La flama de AA usa una hendidura de tipo mechero para incrementar la longitud de la trayectoria y así incrementar la absorbancia total. Las muestras líquidas son aspiradas por un flujo de gas hacia una cámara de nebulación/combinación para formar gotas pequeñas antes de entrar a la flama.
El horno de grafito tiene varias ventajas sobre la flama. Es mucho más eficiente y puede aceptar directamente muestras muy pequeñas para usarse directamente. Además produce un ambiente de reducción para así oxidar fácilmente los elementos. Las muestras son puestas directamente el horno y el horno es calentado eléctricamente en varios pasos para secar la muestra, en cenizas de materia orgánica y vaporizar los átomos de analito.
La diferencia más importante entre un espectrofotómetro de absorción atómica y uno de absorción molecular es la necesidad de convertir el analito en átomos libres. El proceso de convertir el analito en sólido, líquido o solución a un átomo gaseoso libre se llama atomización. En la mayoría de los casos la muestra contiene el analito sufre una preparación para dejar el analito en solución orgánica o acuosa. Dos métodos generales de atomización son usados: atomización a la flama y atomización electrotérmica. Poco elementos son atomizados usando otras técnicas.
Atomización a la flama. En la atomización a la flama la muestra es primero convertida en una suspensión de gotitas de la solución. Esto se logra usando un ensamble nebulizador. La muestra es aspirada en una cámara de spray pasando un vapor a alta presión que consiste de uno o más gases de combustión, pasa al final de un tubo capilar sumergido en la muestra. El impacto de la muestra con el vidrio produce gotas en una solución en aerosol. La suspensión en aerosol se combina con los gases de combustión en la cámara de spray antes de pasar al quemador donde la energía térmica de la flama desolvata la suspensión de aerosol para secar el aerosol a pequeñas, partículas sólidas. Después, la energía térmica volatiliza las partículas, produciendo vapor que consiste de las especies moleculares, especies iónica y los átomos libres.
La energía térmica en la atomización a la flama es suministrada por la combinación de una mezcla combustible oxidante. Los combustibles comúnmente usados son aire-acetileno y óxido de nitrógeno-acetileno. Normalmente, el combustible y el oxidante son mezclados en proporciones estequiométricas; sin embargo, una mezcla rica puede ser aceptable para que los átomos sean fácilmente oxidables. El diseño más común para el quemador es con una ranura. Este quemador provee una longitud de la trayectoria para monitorear la absorbancia y una flama estable.
El quemador es montado en una fase ajustable que permite al quemador ensamblarse para moverse vertical y horizontalmente. El ajuste horizontal es necesario para asegurarse que la flama está alineada con la trayectoria de los instrumentos ópticos. El ajuste vertical es necesario para ajustar la altura dentro de la flama en el que la absorbancia es monitoreada. Esto es importante porque dos procesos que compiten, afectan la concentración de los átomos libres. Un incremento en la residencia del tiempo resulta en una mejor eficiencia de la atomización; entonces la producción de átomos libres se incrementa con la altura. Por otro lado, una residencia muy larga de tiempo, puede conducir a la formación de óxidos metálicos, como Cr, la concentración de los átomos libres es más grande en la cabeza del quemador. Para metales como Ag, que son difíciles de oxidar, la concentración de los átomos libres se incrementa firmemente con la altura. Otros átomos muestran perfiles de concentración que se maximizan a las características de la altura.
La manera más común de introducir la muestra en el atomizador de flama es por continua aspiración, en el cual la muestra es pasada continuamente a través del quemador mientras se monitora la absorbancia. La continua aspiración de la muestra, requiere de 2-5 ml de muestra. Se puede también alimentar micro-muestras que es útil cuando el volumen es limitado o cuando la matriz de la muestra no es compatible con el atomizador de flama. Por ejemplo, la continua aspiración de muestra que contiene altas concentraciones de sólidos disueltos, como agua de mar, puede resultar en la acumulación de depósitos de sólidos en la cabeza del quemador. Estos depósitos generalmente obstruyen la flama, bajando la absorbancia. La inmersión de la muestra se logra con un muestreador automático. Las alimentación de las micro-muestras a la flama se logra usando una micro-pipeta para poner 50-250 ðl de muestra en un embudo de teflón conectado al nebulizador, o sumergiendo el tubo nebulizador en la muestra por corto tiempo. La sumersión de la muestra se logra con un muestrador automático. La señal para micro-muestras es un pico transitorio en el que su altura o área es proporcional a la cantidad de analito que es inyectado.
La principal ventaja de la flama por atomización es la reproducibilidad con que la muestra es inyectada en el espectrofotómetro. Una desventaja significante que la eficiencia de la atomización puede ser muy pobre. Esto puede ocurrir por dos razones. Primero, la mayoría del aerosol producido durante la nebulización consiste de gotas que son muy grandes para ser acarreadas hacia la flama por los gases de combustión. Consecuentemente, casi el 95% de la muestra nunca llega a la flama. La segunda razón es que un volumen grande de gases de combustión significativamente diluye la muestra. Juntas, estas contribuciones a la eficiencia de atomización pueden reducir la sensibilidad, si la concentración del analito en la flama, es de 2.5x10-6 en esa solución.
Atomizadores electrotérmicos.
Una significativa mejora en la sensibilidad se logró con el calentamiento por resistividad en lugar de la flama. Un atomizador electrotérmico muy común, es conocido como horno de grafito, que consiste de un tubo cilíndrico de grafito de aproximadamente 1-3 cm de longitud, y 3-8 mm de diámetro. El tubo de grafito es alojado en un ensamble que sella las salidas del tubo con ventanas ópticamente transparentes. El ensamble también permite el paso de corrientes de gas inerte, protegiendo el grafito de la oxidación, y removiendo los productos gaseosos producidos durante la atomización. Una fuente de poder es usada para pasar la corriente a través del tubo de grafito, resultando en un calentamiento por la resistencia.
Las muestras entre 5 y 50 ðl, son inyectadas al tubo de grafito a través de un hoyo de diámetro pequeño localizado en la parte superior del tubo. La atomización se logra en tres fases. Primero, la mezcla es secada usando una corriente que eleva la temperatura del tubo de grafito a 110ºC. En la segunda etapa, que se llama calcinado, la temperatura es incrementada a 350-1200ºC, a estas temperaturas cualquier material orgánico es convertido en CO2 y H2O, y materiales inorgánicos son volatilizados. Estos gases son removidos por una corriente de gas inerte. En la etapa final, la muestra es atomizada rápidamente incrementando la temperatura a 2000-3000ºC. El resultado es un pico perecedero cuya altura o área es proporcional a la cantidad de analito inyectado en el tubo. Estas tres etapas se llevan a cabo en 45-90 segundos, la mayor parte del tiempo es usada para secar y calcinar la muestra.
La atomización electrotérmica provee una significativa mejora en la sensibilidad atrapando el analito gaseoso en un pequeño volumen en el tubo de grafito. La concentración del analito resultante en el vapor puede ser 1000 veces más grande que la producida en la atomización a la flama. El avance en sensibilidad y en la detección de límites, es compensado por una significativa pérdida en la precisión. La eficiencia de la atomización está fuertemente influenciada por el contacto de la muestra con el tubo de grafito, en el que es difícil controlar la reproducibilidad.
Métodos de atomización misceláneos.
Pocos elementos pueden ser atomizados por una reacción química que produce un producto volátil. Elementos como As, Se, Sb, Bi, Ge, Sn, Te y Pb forman hidruros volátiles tanto a la flama como en una observación de un tubo de cuarzo calentado en la trayectoria óptica. El Hg es determinado por un método de vapor frío en el que es reducido a mercurio elemental con SnCl2. El mercurio volátil es acarreado por un gas inerte a un tubo de observación situado en la trayectoria óptica del instrumento.
FUNDAMENTO TEÓRICO DE LA TÉCNICA.
La ley de Lambert-Beer (también llamada ley de Beer-Lambert-Bouguer) es una relación lineal entre la absorbancia y la concentración de la absorción de la radiación electromagnética. La fórmula general de la ley es:
A=aðxbxc
Donde A es la absorbancia transmitida, a una ð longitud de onda que depende del coeficiente de absorción, b es la longitud de la trayectoria, y c es la concentración del analito. Cuando la concentración se maneja en molaridad, la ley de Lambert-Beer se escribe:
A=ððxbxc
Donde ðð es la longitud que depende de la coeficiente de absorción molar en unidades M-1cm-1. La ð de subíndice se sobreentiende como un valor específico de para una longitud de onda dada. Si varias especies absorben luz a una longitud de onda en una muestra, la absorbancia total a la longitud de onda dada es:
A=(ððxbxc1)+ (ððxbxc2)+....
Donde los subíndices se refieren a absortividad y la concentración de las diferentes sustancias que están presentes.
Mediciones experimentales son generalmente hechas en términos de transmitancia (T) que es definida por:
donde P es la potencia de la luz a través de la muestra y Po es la potencia inicial de la luz, la relación entre A y T es:
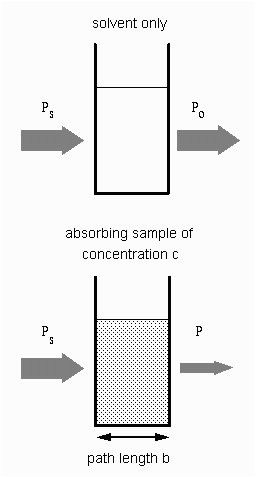
En aplicaciones analíticas queremos medir la concentración de analito independiente de los efectos de la reflexión, absorción del solvente, y otras interferencias. La figura de la derecha muestra las dos mediciones de transmitancia que son necesarias para usar la absorción y así calcular la concentración del analito en solución. El diagrama de arriba es solo para el solvente y el de abajo es para la absorción y solvente de la muestra. En este ejemplo, Ps es la potencia de la luz que incide en la muestra, P es la potencia de la luz medida después de que pasa el analito, el solvente y el contenedor de la muestra (celda) y Po es la potencia de la luz medida después que atraviesa el analito solamente y la celda. La medida de la transmitancia en este caso se le atribuye al analito únicamente.
Dependiendo del tipo de instrumento, la medida de referencia puede ser hecha simultáneamente con la medición de la muestra o con la referencia gravada en la computadora para generar el espectro completo.
Los modernos aparatos de absorción pueden mostrar los datos en transmitancia, % de transmitancia, o absorbancia. Una concentración desconocida de analito puede ser determinada con la medición de la cantidad de luz que absorbe la muestra aplicando la ley de Beer. Si el coeficiente de absortividad no se conoce, la concentración se puede determinar usando una curva de calibración de la absorbancia contra la concentración derivadas de los estándares.
Limitaciones de la ley de Lambert-Beer.
La linearidad de la ley de Lambert-Beer es limitada por los factores químicos o instrumentales. Las causas de la no-linearidad son:
-
Desviaciones de los coeficientes de absorbancia a altas concentraciones (>0.01M) debido a las interacciones electrostáticas entre las moléculas por la proximidad.
-
Dispersión de la luz debido a las partículas de la muestra
-
Fluorescencia o fosforescencia de la muestra
-
Cambios en la índice de refracción debido a las altas concentraciones
-
Cambios en el equilibrio químico como función de la concentración
-
Radiación no monocromática, las desviaciones pueden ser minimizadas usando una parte uniforme del espectro de absorción como el máximo de absorción de banda
-
Desviación de la luz.
APLICACIÓN DE LA TÉCNICA.
Molybdenum at Germany Norway spruce sites: Contents and mobility
Canadian Journal of Forest Research; Ottawa; Jul 2000; Friederike Lang; Martin Kaupenjohann;
| Foreign Title: | Revue canadienne de recherche forestiere |
| Volume: | 30 |
| Issue: | 7 |
| Start Page: | 1034 |
| ISSN: | 00455067 |
| Full Text: | |
| Copyright National Research Council of Canada Jul 2000 | |
[Headnote]
Abstract: El molibdeno juega un papel importante en el ciclo del nitrógeno en los ecosistemas. Sin embargo, se sabe muy poco de la disponibilidad de Mo es los suelos de los bosques. Medimos el oxalato extraíble de la concentración de Mo en las tierras ácidas de los bosques, de los flujos de Mo, nitrato, fosfato del suelo orgánico en los minerales de la tierra usando tubos de resina y las concentraciones de Mo de las tres agujas en 28 diferentes piceas noruegas. El suministro de oxalato-extraíble de Mo varió de 51 a 3400 g/ha, con los valores más bajos que ocurrieron en la piedra arenisca derivada de la tierra (370 +/- 212 g/ha; mean +/- SD). Las concentraciones de molibdeno en el presente año estuvieron en un rango de 5 a 48ng/g. Las concentraciones de la aguja y el oxalato extraíble de Mo no se correlacionaron. Sin embargo, los flujos de Mo (6-60 g/ha*a) de los suelos de tierra orgánica en los minerales estuvieron correlacionados con las concentraciones de las agujas y los flujos de NO3. Concluimos que el ciclo de Mo en los ecosistemas de los bosques están gobernados por la habilidad de las sales minerales de Mo así como también la adaptación de la planta al Mo. Además, el ciclo de Mo afecta fuertemente la distribución de Mo dentro de los perfiles de tierra y los flujos de Mo fuera de las capas orgánicas.
Objetivo: investigar porque la concentración de molibdeno ha estado bajando en las agujas de las piceas en Alemania y qué está relacionada dicha concentración. Las plantas que exhiben una carencia de molibdeno muestran bajas concentraciones de nitrógeno, a pesar de que las plantas crecen con grandes soluciones de NO3. Tratar de comprender los bajos niveles de nitrógeno si hay grandes cantidades de NO3.
Resumen: para explicar estos fenómenos, se explicó el muestreo de la tierra y de las agujas para obtener las muestras. Aclaró cómo se prepararon las muestras para su análisis. Para medir las concentraciones de molibdeno se uso espectroscopia de absorción atómica. Otros metales determinados con la misma técnica fueron el Fe y Al. El espectrómetro usado fue SpectrAA 800 Z, Varian. Otras técnicas que se usaron, fueron la cromatografía de gases y colorimetría.
Conclusiones: se hicieron inferencias estadísticas para llegar a las conclusiones. Los resultados muestran que los suministros de molibdeno están determinados por los minerales padres de la tierra. En contraste las concentraciones en las capas superiores de tierra se deben al ciclo del molibdeno dentro del sistema, sin consideración a los materiales padre. Se hipotetiza que la planta toma el molibdeno de las capas orgánicas. La equidad entre el molibdeno y el nitrato sugiere que las grandes concentraciones de nitrato, ayudan a la fijación del molibdeno.
EJEMPLOS ADICIONALES.
-
Espectroscopia de absorción atómica en productos alimenticios para cursos de carreras no científicas.
-
Detección de la cantidad de excreción de oxalato y calcio en la fibrosis cística.
-
Estructura las minúsculas partículas de los cristales de bario-titanio incluyendo magnesio y el análisis de su vibración de red.
Bibliografía:
Modern Analytical Cehmistry. David Havey. Mc. Fraw Hill, 2000. USA. Pp 412-418.
Descargar
| Enviado por: | Eduardo Mosso |
| Idioma: | castellano |
| País: | México |
Todos los derechos reservados.