Biología, Botánica, Genética y Zoología
Anomalías cromosómicas en número y estructura
ANOMALÍAS CROMOSÓMICAS
INTRODUCCION
Cualquier alteración en el número y/o en la morfología de los cromosomas constituye una alteración cromosómica.
Cuando existen uno o más juegos de cromosomas completos, se habla de euplodía (triploidía, tetraploidía y en general poliploidía). En el caso de existir un defecto de cromosomas, se habla de monosomía. Si el defecto o exceso es de cromosomas incompletos, se habla de aneuploidias
Las alteraciones estructurales se refieren a cambios en la forma y/o tamaño de un cromosoma. Cuando el material genético se conserva en el cromosoma alterado, la alteración es equilibrada, mientras que si se gana o pierde material genético, la alteración es desequilibrada. Son la consecuencia de la rotura y uniones anómalas de los cromosomas bajo la influencia de agentes externos que la célula no puede reparar. Las alteraciones estructurales básicas son las roturas que ocasionan bien la formación de una deleción (cromosoma al que le falta un fragmento) o de un fragmento sin centrómero
Casi la mitad de las alteraciones cromosómicas que se encuentran en el recién nacido son la presencia de un cromosoma extra (aneuploidía) ya que las monosomías totales son incompatibles con la vida. Las trisomías constituyen la anomalía cromosómica más frecuente y, dentro de estas, las más conocidas son la trisomía 21 (síndrome de Down), la trisomía 18 (síndrome de Edwards) y la trisomía 13 (síndrome de Patau). Solo los niños con síndrome de Down sobreviven hasta la edad adulta, mientras que los que tienen trisomías 18 y 13 mueren por lo general antes del primer año.
Los efectos de estas alteraciones se estudian a diario en diferentes laboratorios del mundo con el fin de encontrar la solución a esos defectos y brindar una mejor calidad de vida a aquellos individuos que se ven afectados por las diferentes enfermedades.
Uno de las mas grandes investigaciones que se han llevado a cabo fue la del PROYECTO GENOMA hace mas de dos años en el que descifraban cada uno de los cromosomas humanos y los síndromes o enfermedades que causaban en caso de presentar una anomalía.
Nuestro propósito es conocer las diferentes anomalías tanto numéricas como cromosomicas en humanos y animales, aunque los resultados se mostraron mas en humanos…
DESCRIPCIÓN GENERAL DE LAS ANOMALÍAS CROMOSÓMICAS
¿Qué es un cromosoma?
El cuerpo humano está constituido de células. Por ejemplo, cuando una persona sufre una quemadura solar, la piel se desprende y deja caer "células" de la piel. En el centro de cada célula existe un área llamada núcleo. Los cromosomas humanos se encuentran en el núcleo de la célula. El cromosoma es una estructura que forma parte del núcleo y que contiene los genes. Los genes determinan los rasgos, como el color de ojos y el grupo sanguíneo.
¿Cómo se heredan los cromosomas?
Normalmente, cada célula de nuestro cuerpo tiene un total de 46 cromosomas, o 23 pares. Heredamos la mitad de los cromosomas (un miembro de cada par) de nuestra madre biológica y la otra mitad (el miembro homólogo de cada par) de nuestro padre biológico.
Los científicos han enumerado los pares de cromosomas de 1 a 22, habiéndole dado al par 23 el nombre de X o Y, según la estructura. Los primeros 22 pares de cromosomas se llaman "autosomas". Los cromosomas del par 23 se conocen como los "cromosomas sexuales" porque determinan si el bebé será varón o mujer. Las mujeres tienen dos cromosomas "X" y los hombres tienen un cromosoma "X" y un cromosoma "Y". La representación gráfica de los 46 cromosomas, ordenados en pares, recibe el nombre de cariotipo. El cariotipo normal de la mujer se escribe 46, XX, mientras que el cariotipo normal del hombre se escribe 46, XY.
LAS ANOMALÍAS NUMÉRICAS: DESCRIPCIÓN GENERAL DE LAS TRISOMÍAS Y LAS MONOSOMÍAS
¿Qué son las anomalías cromosómicas numéricas?
Las anomalías numéricas conforman uno de los tipos de anomalías cromosómicas. Estos tipos de defectos congénitos ocurren cuando hay un número de cromosomas diferente en las células del cuerpo que el número normal. De modo que, en lugar de los 46 cromosomas habituales en cada célula del cuerpo, hay 45 ó 47 cromosomas. El tener demasiados cromosomas o una cantidad insuficiente de cromosomas constituye una causa para el desarrollo de algún defecto congénito.
¿Qué son las trisomías?
El término "trisomía" se utiliza para describir la presencia de tres cromosomas en lugar del par habitual de cromosomas. Por ejemplo, si un niño nace con tres cromosomas 21 en lugar del par usual, se diría que posee "trisomía 21". La trisomía 21 también se conoce como síndrome de Down. Otros ejemplos de trisomía incluyen la trisomía 18 y la trisomía 13. Nuevamente, trisomía 18 o trisomía 13 significa simplemente que existen tres copias y no el par usual del cromosoma 18 (o del cromosoma 13) en cada célula del cuerpo.
¿Qué son las monosomías?
El término "monosomía" se utiliza para describir la ausencia de un miembro de un par de cromosomas. Por lo tanto, habrá un total de 45 cromosomas en cada célula del cuerpo, en lugar de 46. Por ejemplo, si un bebé nace con un solo cromosoma sexual X, en lugar del par habitual (ya sea, dos cromosomas sexuales X o un cromosoma sexual X y un cromosoma sexual Y), se dirá que tiene "monosomía X." La monosomía X también se conoce con el nombre de síndrome de Turner.
EL SÍNDROME DE DOWN (TRISOMÍA 21)
¿Qué es el síndrome de Down?
El síndrome de Down es un trastorno genético que implica una combinación de defectos congénitos, entre los que se incluyen cierto grado de retardo mental, rasgos faciales característicos y, a menudo, defectos cardíacos, deficiencia visual y auditiva y otros problemas de salud. La gravedad de todos estos problemas varía en gran medida entre los individuos afectados. Este síndrome es uno de los defectos genéticos congénitos más comunes y afecta aproximadamente a uno cada 800 a 1000 niños. Según el Instituto Nacional de Síndrome de Down (National Down Syndrome Society), existen más de 350.000 individuos que padecen este síndrome en Estados Unidos. La expectativa de vida de adultos con síndrome de Down es de 55 años aproximadamente, aunque el período de vida promedio varía.
El nombre "síndrome de Down" proviene del médico Dr. Langdon Down, quien fue el primero en describir el conjunto de descubrimientos en 1866. No fue sino hasta 1959 que se identificó la causa del síndrome de Down (la presencia de un cromosoma 21 adicional).
¿Cuáles son las causas del síndrome de Down?
Normalmente en la reproducción, el óvulo de la madre y el espermatozoide empiezan teniendo el número usual de 46 cromosomas. El óvulo y el espermatozoide sufren una división celular en donde los 46 cromosomas se dividen en dos partes iguales y el óvulo y el espermatozoide finalmente poseen 23 cromosomas cada uno. Cuando un espermatozoide con 23 cromosomas fertiliza un óvulo con 23 cromosomas, el bebé tiene finalmente un grupo completo de 46 cromosomas, una mitad obtenida del padre y la otra mitad de la madre.
A veces, ocurre un error mientras los 46 cromosomas se dividen a la mitad y el óvulo o el espermatozoide, en lugar de reservar tan solo una copia del cromosoma 21, sigue teniendo ambas. Si este óvulo o espermatozoide se fertiliza, el bebé acabará teniendo tres copias del cromosoma 21 y esto es lo que se llama "trisomía 21" o síndrome de Down. Las características del síndrome de Down se originan porque cada célula del cuerpo posee una copia adicional del cromosoma 21.
El noventa y cinco por ciento de los casos de síndrome de Down se produce por la Trisomía 21. En algunas ocasiones, el cromosoma 21 adicional se adhiere a otro cromosoma del óvulo o el espermatozoide; esto puede conducir a lo que se denomina síndrome de Down por "translocación" (el 3 a 4 por ciento de los casos). Éste es el único tipo de síndrome de Down que puede, a veces, heredarse de alguno de los padres. Algunos padres tienen un reordenamiento que no afecta su salud denominado translocación balanceada, donde el cromosoma 21 se adhiere a otro cromosoma. Con poca frecuencia, tiene lugar un tipo de síndrome de Down llamado el síndrome de Down con alteración cromosómica en "mosaico", cuando ocurre un error en la división celular después de la fertilización (1 a 2 por ciento de los casos). Estas personas tienen algunas células con un cromosoma 21 adicional y otras con el número normal.
LA TRISOMÍA 18 Y 13
¿Qué es la trisomía 18 y la trisomía 13?
La trisomía 18 y la trisomía 13 son trastornos genéticos que presentan una combinación de defectos congénitos que incluyen retardo mental grave, así como problemas de salud que comprometen a casi todos los sistemas orgánicos del cuerpo. Entre el 20 y el 30 por ciento de los bebés que nacen con trisomía 18 o trisomía 13 mueren durante el primer mes de vida, y el 90 por ciento muere al año. Es importante destacar que entre el 5 y el 10 por ciento de los bebés con trisomía sobreviven al primer año de vida. Por lo tanto, estos trastornos no son fatales en todos los casos y, ante la ausencia de problemas inmediatos que pongan en peligro la vida, es difícil hacer predicciones precisas respecto de la expectativa de vida. Existen algunos informes sobre bebés con trisomía 18 o 13 que sobrevivieron hasta la adolescencia. Sin embargo, estos casos son poco frecuentes.
La trisomía 18 se denomina también "síndrome de Edwards", llamada así por el primer médico que describió el trastorno. La trisomía 18 se observa en aproximadamente uno de cada 5.000 a 10.000 recién nacidos vivos.
La trisomía 13 se denomina también "síndrome de Patau", llamada así por el primer médico que describió el trastorno. La trisomía 13 se observa en aproximadamente uno de cada 5.000 recién nacidos vivos.
¿Cuáles son las causas de la trisomía 18 y la trisomía 13?
En general cada óvulo y cada espermatozoide contiene 23 cromosomas. La unión de estos crea 23 pares, o 46 cromosomas en total, cuando se realiza la fecundación. De esta manera, una persona recibe exactamente la mitad de su material genético de cada uno de los padres. En ocasiones, ocurre un error durante la formación del óvulo o del espermatozoide, y esto causa la presencia de un cromosoma 18 o 13 adicional. Cuando esta célula aporta el cromosoma 18 adicional al embrión, el resultado es la trisomía 18. Cuando esta célula otorga el cromosoma 13 adicional al embrión, el resultado es la trisomía 13. El cromosoma 18 o 13 adicional puede provenir tanto del óvulo de la madre como del espermatozoide del padre. Las características de la trisomía 18 y la trisomía 13 son el resultado de la presencia de este cromosoma 18 o 13 adicional en cada célula del cuerpo.
En algunas ocasiones, el cromosoma 18 o 13 adicional se adhiere a otro cromosoma en el óvulo o el espermatozoide; esto se denomina translocación. ésta es la única forma de trisomía 18 o 13 que puede ser hereditaria. A veces, uno de los padres puede portar un reordenamiento "balanceado" en el cual el cromosoma 18 o 13 se adhiere a otro cromosoma. Sin embargo, como el padre no tiene ningún material cromosómico adicional o ausente, se dice que presenta una "translocación balanceada" y suele ser normal y goza de buena salud. Esporádicamente, puede ocurrir una trisomía 18 o 13 en mosaico cuando el error en la división celular ocurre después de la fecundación. Estas personas poseen algunas células con un cromosoma 18 o 13 adicional y otras con la cantidad normal.
EL SÍNDROME DE TURNER
¿Qué es el síndrome de Turner?
El síndrome de Turner es un trastorno genético que se presenta en las niñas y que provoca que sean más bajas que el resto y que no maduren sexualmente a medida que alcanzan la edad adulta. La gravedad de estos problemas varía entre los individuos afectados. También pueden presentarse otros problemas de salud que comprometen al corazón o al aparato renal (es decir, los riñones). Muchos de los problemas que afectan a las niñas con síndrome de Turner pueden controlarse o corregirse con el tratamiento médico adecuado. Este síndrome afecta a una de cada 2.500 niñas recién nacidas.
El nombre "síndrome de Turner" proviene del médico Dr. Henry Turner, quien fue el primero en describir el conjunto de descubrimientos en 1938. No fue sino hasta 1959 que se identificó la causa del síndrome de Turner (la presencia de un sólo cromosoma X).
¿Cuáles son las causas del síndrome de Turner?
Normalmente en la reproducción, el óvulo de la madre y el espermatozoide del padre comienzan teniendo el número habitual de 46 cromosomas. El óvulo y el espermatozoide sufren una división celular en donde los 46 cromosomas se dividen en dos partes iguales y el óvulo y el espermatozoide poseen finalmente 23 cromosomas cada uno. Cuando un espermatozoide con 23 cromosomas fertiliza un óvulo con 23 cromosomas, el bebé tiene finalmente un grupo completo de 46 cromosomas, una mitad obtenida del padre y la otra mitad de la madre.
En ocasiones, ocurre un error durante la formación del óvulo o del espermatozoide, lo que provoca que éste posea un cromosoma sexual menos. Cuando esta célula no puede otorgar el cromosoma sexual al embrión, de manera que existe sólo un cromosoma sexual X, el resultado es el síndrome de Turner. El hecho de tener una sola copia de un cromosoma determinado, en lugar del par habitual, se denomina "monosomía". El síndrome de Turner también se conoce como "monosomía X." El error del cromosoma sexual faltante puede ocurrir tanto en el óvulo como en el espermatozoide; sin embargo, suele ser un error que ocurre en la formación del espermatozoide. No existe nada conocido que el padre pueda haber hecho o no que pudiera haber causado o prevenido la falta de un cromosoma sexual en la formación del espermatozoide. (La probabilidad de que aparezca el síndrome de Turner por lo tanto, no se asocia con la edad de la madre). Las características de este síndrome se originan por la falta de un cromosoma X en cada una de las células del cuerpo.
Alrededor del 50 por ciento de los casos de síndrome de Turner resulta de la monosomía X total. El cuarenta por ciento de los casos son "en mosaico", lo que significa que estas personas tienen algunas células sin un cromosoma X y otras con la cantidad normal de cromosomas. Un por ciento pequeño del síndrome de Turner son el resultado de una cantidad normal de cromosomas (46 en total), pero con la falta de una porción del cromosoma X. Cuando falta sólo una parte del cromosoma X (denominado deleción), no todo el cromosoma, las niñas con síndrome de Turner suelen tener características menos pronunciadas del síndrome. Las características del síndrome de Turner presentes dependen de la parte faltante del cromosoma X.
LAS ANOMALÍAS ESTRUCTURALES: LAS DELECIONES (CRI DU CHAT) Y LAS DUPLICACIONES (PALLISTER KILLIAN)
¿Cuáles son las anomalías cromosómicas estructurales?
Las anomalías cromosómicas estructurales se presentan cuando hay un cambio en la estructura o en los componentes de un cromosoma. Generalmente el total de cromosomas es normal (46 por célula). Las anomalías cromosómicas estructurales ocurren cuando se pierde parte del cromosoma, cuando hay material cromosómico adicional o cuando dos partes se han intercambiado de lugar. Como consecuencia, esto conduce al exceso o a la carencia de material genético, lo que provoca algunos defectos congénitos.
Cada cromosoma está constituido por varios segmentos que generalmente se clasifican como "brazo corto" y "brazo largo". El brazo corto, la mitad superior del cromosoma, se denomina "brazo p" y el brazo largo, la mitad inferior del cromosoma, es el "brazo q". El centrómero es la parte de centro de un cromosoma que aparezca "pellizcado" entre los brazos "p" y "q".
¿Qué son las deleciones?
El término "deleción" significa simplemente que una parte del cromosoma se perdió o se "eliminó". Un pieza muy pequeña de un cromosoma puede contener muchos genes diferentes. Cuando hay pérdida de material genético, puede haber errores en el desarrollo del bebé, como consecuencia de la pérdida de algunas de las "instrucciones". Un ejemplo de un síndrome genético provocado por deleción es el denominado "Cri du Chat", en el cual ocurre una deleción o pérdida de parte del cromosoma 5.
¿Qué es el Cri du Chat?
Anualmente, el Cri du Chat o "síndrome del maullido de gato" se encuentra en aproximadamente uno en 20.000 a 50.000 nacimientos vivos en los Estados Unidos. La causa del Cri du Chat es la deleción del cromosoma 5p, cuya nomenclatura es "5p-". Las características de los bebés que sufren Cri du Chat son: llanto muy agudo, falta de tonicidad muscular, microcefalia y bajo peso al nacer. También tienen problemas con el lenguaje, y es posible que se expresen a través de una cantidad reducida de palabras o lenguaje de señas. Otros problemas de salud que pueden presentarse incluyen retardo para comenzar a caminar, problemas en la alimentación, hiperactividad, escoliosis y retardo mental severo. El promedio de vida de la mayoría de las personas con Cri du Chat estará dentro de lo normal, a menos que nazcan con otros defectos severos en los órganos.
Para lograr el desarrollo pleno del potencial de un niño con Cri du Chat, es importante la educación desde una edad temprana, además de terapias físicas y del lenguaje.
¿Qué son las duplicaciones?
El término "duplicación" significa simplemente que una parte del cromosoma está duplicada o presenta dos copias. El resultado es el material genético adicional, aun cuando el total de cromosomas está generalmente dentro de lo normal. Dado que una pieza muy pequeña de un cromosoma puede contener muchos genes diferentes, el material genético presente en una duplicación puede provocar que dichos genes no funcionen correctamente. Como consecuencia de estas "instrucciones adicionales", pueden producirse errores en el desarrollo de un bebé. Una manera de pensar en la duplicación es pensar que los 46 cromosomas forman un libro de cocina, y cada uno de los cromosomas es una receta. Si una deleción es un ingrediente que falta en una receta, un duplicación es un ingrediente adicional. Un ejemplo de un síndrome genético provocado por duplicación es el denominado "síndrome de Pallister Killian", en el cual parte del cromosoma 12 está duplicado.
Este nuevo cromosoma que se forma se denomina cromosoma por translocación. La translocación de este ejemplo se encuentra entre los cromosomas 14 y 21. Cuando un bebé nace con este tipo de cromosoma por translocación (entre el 14 y el 21), además de un cromosoma 14 normal y dos cromosomas 21 normales, el bebé sufrirá síndrome de Down, también denominado síndrome de Down por translocación.
¿Qué es el síndrome de Pallister Killian?
El síndrome de Pallister Killian es el resultado del material genético adicional del cromosoma 12. En general, se presenta una mezcla de células (mosaicismo), algunas con material adicional del cromosoma 12 y otras normales (46 cromosomas sin material genético adicional del cromosoma 12). Los bebés que sufren este síndrome padecen muchos problemas, entre los que se incluyen retardo mental severo, falta de tonicidad muscular, facciones toscas y frente prominente. Entre otras características se pueden mencionar el labio superior muy delgado, el labio inferior más grueso y la nariz corta. Entre otros problemas de salud que ocasiona este síndrome se incluyen convulsiones, mala alimentación, rigidez en las articulaciones, cataratas (en los adultos), pérdida auditiva y defectos cardíacos. El promedio de vida de las persona que padecen síndrome de Pallister Killian es reducido, aunque pueden vivir más de 40 años.
LAS TRANSLOCACIONES
¿Qué son las translocaciones?
El término translocación se utiliza cuando se presentan modificaciones en la ubicación de determinado material cromosómico. Existen dos tipos de translocaciones: recíproca y robertsoniana. En una translocación recíproca, dos cromosomas diferentes intercambian segmentos entre sí.
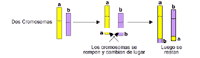
En una translocación robertsoniana, un cromosoma completo se adhiere a otro en el centrómero. El centrómero es la parte de centro de un cromosoma que aparezca "pellizcado" entre los brazos "p" y "q".

Este nuevo cromosoma que se forma se denomina cromosoma por translocación. La translocación de este ejemplo se encuentra entre los cromosomas 14 y 21. Cuando un bebé nace con este tipo de cromosoma por translocación (entre el 14 y el 21), además de un cromosoma 14 normal y dos cromosomas 21 normales, el bebé sufrirá síndrome de Down, también denominado síndrome de Down por translocación.
EL SÍNDROME DE DOWN POR TRANSLOCACIÓN
¿Qué es el síndrome de Down por translocación?
El síndrome de Down por translocación hace referencia al reordenamiento del material cromosómico. Existen tres cromosomas 21, al igual que en la trisomía 21, pero uno de ellos está adherido a otro cromosoma, en lugar de estar separado. El cromosoma 21 adicional es el que provoca los problemas que constituyen el síndrome de Down. En el síndrome de Down por translocación, el cromosoma 21 adicional puede adherirse al cromosoma 14, o al 13, 15 o 22. En algunos casos, dos cromosomas 21 pueden adherirse uno a otro.
Entre el 3 y el 4 por ciento de los bebés que nacen con síndrome de Down tienen síndrome de Down por translocación. En una habitación con 100 bebés con síndrome de Down, todos los bebés se parecerán entre sí y tendrán características y problemas de salud similares. No será fácil detectar los 3 o 4 que presenten la translocación.
Cada vez que se encuentra una translocación en un niño, se estudian los cromosomas de los padres para determinar si dicha translocación es hereditaria o no. Si uno de los padres presenta un cromosoma por translocación, entonces el médico concluye que el bebé heredó ese cromosoma de su padre. En realidad, el padre tendrá un total de 45 cromosomas en cada célula de su cuerpo, cuyo estado será normal y saludable debido a que aún tienen sólo dos copias de cada cromosoma. Cuando una persona presenta un reordenamiento del material cromosómico, sin material cromosómico extra o ausente, se dice que tiene una "translocación balanceada" o que es un "portador de translocación balanceada".
Los padres con translocaciones balanceadas pueden presentar problemas de fertilidad (problemas para gestar), abortos espontáneos o mayor probabilidad de tener un hijo con problemas de salud. Aunque el padre puede donar la cantidad adecuada de material genético (23 cromosomas) para lograr el embarazo, corre el riesgo de que este material donado sea demasiado o insuficiente. Esto no puede controlarse ni predecirse. Las probabilidades dependen del tipo de reordenamiento cromosómico y de los cromosomas involucrados. Por ejemplo, si la translocación está entre los cromosomas 14 y 21, las probabilidades de síndrome de Down en el embarazo son de entre un 10 y un 15 por ciento si la madre es la portadora de la translocación, y de entre un 3 y un 5 por ciento si el padre es el portador de la translocación. Las probabilidades son diferentes para hombres y mujeres debido a que los espermatozoides y los óvulos se producen de manera diferente. La mujer nace con todos los óvulos que tendrá a lo largo de su vida, mientras que el hombre produce espermatozoides nuevos constantemente.
Existe otro factor importante a tener en cuenta cuando se encuentra una translocación en uno de los padres. Los familiares de los padres (hermanos, hermanas) también pueden haber heredado la translocación y, por lo tanto, pueden correr los mismos riesgos en un embarazo. Por estos motivos, se recomienda que las personas con reordenamientos cromosómicos compartan esta información con sus familiares para que estos puedan realizarse los estudios correspondientes.
OTROS ARREGLOS CROMOSÓMICOS: ANILLOS E INVERSIONES
¿Qué es una inversión?
Existen diversas formas en las que puede alterarse la estructura de un cromosoma. Una de ellas se denomina "inversión". El término "inversión" se utiliza cuando un cromosoma se fragmenta en dos puntos y el segmento intermedio gira al revés y luego vuelve a unirse.
Algunas inversiones son tan frecuentes en la población que no es necesario realizar una prueba adicional para detectarlas. Si se encuentra una inversión rara o poco frecuente en un niño, se estudian los cromosomas de los padres para determinar si dicha inversión es hereditaria o no. Si uno de los padres presenta esta inversión, entonces el médico concluye que el bebé heredó dicha inversión de su padre. El hecho de que generalmente el estado del padre sea normal y saludable prueba que la inversión no provocó ningún daño genético en el momento en que el cromosoma se fragmentó y giró al revés. (Los demás familiares de los padres también pueden haber heredado la inversión y pueden decidir si realizar o no un estudio cromosómico.) Sin embargo, si la inversión no se presenta en alguno de los padres, existe la posibilidad de que parte del material genético se haya perdido o alterado en el reordenamiento. Esto podría provocar problemas en el desarrollo del bebé, incluyendo defectos congénitos.
¿Qué es un cromosoma anular?
Otro ejemplo de reordenamiento cromosómico es lo que se denomina cromosoma "anular". El término cromosoma "anular" se utiliza para describir un cromosoma cuyas extremidades se fragmentaron y se unieron para formar un círculo o "anillo".
Descargar
| Enviado por: | Carlos Benítez |
| Idioma: | castellano |
| País: | Colombia |
Todos los derechos reservados.