Salud
Drepanocitosis
UNIVERSIDAD JUAREZ DEL ESTADO DE DURANGO
FACULTAD DE MEDICINA
GOMEZ PALACIO
HEMATOLOGIA
“DREPANOCITOSIS”
6TO SEMESTRE
SECCION “A”
FECHA DE ENTREGA
11 DE NOVIEMBRE DEL 2004
INTRODUCCIÒN
Componentes de la sangre.
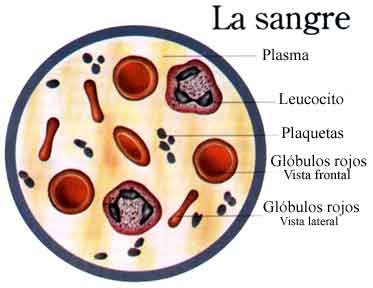
La sangre humana está formada por el plasma sanguíneo, los g1óbulos rojos o eritrocitos, los glóbulos blancos o leucocitos y las plaquetas. Su temperatura es de los 36ºC, y una persona adulta tiene un promedio de unos 5 litros de sangre, lo cual corresponde al 8% del peso de su cuerpo.
El plasma sanguíneo es el componente líquido de la sangre, es decir, una solución que contiene 90-92 % de agua y transporta sus elementos sólidos (glóbulos y plaquetas). Además, presenta una gran variedad de sustancias en disolución, como azúcares, proteínas, grasas, sales minerales, etc. que se pueden agrupar en tres categorías:
• Proteínas: Son albúminas, globulinas y fibrinógeno. El fibrinógeno es el responsable de la formación de coágulos, y la parte de plasma que no lo contiene se denomina suero sanguíneo.
• Sales inorgánicas: Se encuentran disueltas en forma de aniones (iones cloro, bicarbonato, fosfato y sulfato) y cationes (sodio, potasio, calcio y magnesio). Actúan como una reserva alcalina que mantiene constante el pH y regula el contenido de agua.
• Sustancias de transporte: son moléculas que proceden de la digestión (glucosa, aminoácidos) o de la respiración (nitrógeno, oxígeno), residuos del metabolismo (dióxido de carbono, urea, ácido úrico), o bien sustancias absorbidas por la piel, las mucosas, los pulmones, etc.
Los glóbulos rojos o eritrocitos.

Son células de color rojo capaces de captar gran cantidad de oxígeno. En cada milímetro cúbico de sangre existen entre 4,5 a 6 millones. Esta enorme abundancia hace que la sangre tenga un color rojo intenso. Cuando una persona padece de anemia, la cantidad de glóbulos rojos baja de los niveles normales, según la edad y sexo.
Los glóbulos rojos, también denominados eritrocitos o hematíes, son células sanguíneas en forma de disco bicóncavo: un diámetro de 6-9 micras y un espesor de 1 micra, que aumenta progresivamente hacia los bordes (2,2 micras). El ser humano cuenta con 4,5 o 5 millones de eritrocitos por mm3, que constituyen el 45 % del volumen de la sangre.
Los eritrocitos se producen en la médula ósea a partir de una célula madre y mediante un proceso de eritropoyesis. Esta producción es continua porque, cada segundo, los macrófagos del bazo destruyen unos dos millones de hematíes envejecidos que hay que reemplazar.
Se puede considerar que los glóbulos rojos son células «no vivas», ya que carecen de núcleo y de mitocondrias, pero esto no les impide realizar su función: el transporte de oxígeno.
En su interior, los glóbulos rojos están formados básicamente por hemoglobina, una proteína constituida por cuatro cadenas de aminoácidos. Cada cadena se asocia a un grupo molecular, el grupo hemo, cada uno de los cuales cuenta con un átomo de hierro, que fija una molécula de oxigeno y la transporta desde los pulmones hasta los tejidos.
La hemoglobina es uno de los derivados nitrogenados de la ferroprotoporfirina. Es una proteína conjugada que contiene las proteínas básicas incoloras, las globinas y ferroprotoporfirina o hem (el cual consta de una parte orgánica y un átomo de hierro). Esta proteína es la encargada de transportar el O2 en la sangre, por poseer el grupo hem, es similar a la mioglobina.
Estructura Primaria
Las hemoglobinas de todos los mamíferos tienen un peso molecular aproximado de 65.000 y en esencia son tetrámeros, que constan de 4 cadenas péptidas, cada una de las cuales esta unida a un grupo hem. Las moléculas de hemoglobina se forman por combinación de dos subunidades de una cadena peptídica llamada a y dos de b donde las cadenas polipeptídicas están constituidas por eslabones de aminoácidos (AA) denominados residuos; conteniendo 141 residuos la cadena a y 146 la cadena b. Todo ser humano es capaz de sintetizar (genéticamente) e introducir en la hemoglobina cuatro cadenas polipéptidas designadas a, b, g y d. Con escasas excepciones las moléculas de Hb se forman por la combinación de dos cadenas a con dos g o d. La Hb de una persona adulta normal se designa por Hb A = a 2A b 2A y de igual forma, la Hb fetal es Hb A = a 2Ag 2F Las cadenas b, g, d contienen todas ellas 146 unidades que se asemejan mucho entre sí en la secuencia de AA, hay solo 39 residuos de AA diferentes entre las cadenas b y g y solo 10 entre b y d.
Estructura Secundaria
La orientación de las cadenas polipeptídicas puede ser completamente extendida (que no es muy común), por lo que es mejor clasificarlas como a) alfahélice, b) hoja plegada, c) al azar. El porcentaje de contenido de alfahélice en las proteínas globulares es bastante variable (0 - 90%), en el caso de la Hb su contenido es de un 75%. Existen dos factores (o mas bien aminoácidos que pertenezcan a la cadena polipeptídica) que pueden interrumpir la orientación helicoidal: presencia de prolina la cual provoca una torsión de la cadena y, la presencia de fuerzas electrostáticas localizadas de repulsión debido a un conjunto de grupos -R cargados positivamente (lisina y argina), o negativamente (ac- glutámico y aspártico).
En la cadena polipeptídica seleccionada (b 36-59), desde el AA 36 al 42 encontramos una estructura que se forma al enrollarse helicoidalmente sobre si mismo, se debe a la formación de enlaces de hidrógeno entre el -C = O de un AA y el -NH. Del AA 43 al 52, se forma una estructura de hoja plegada, la cual se identifica por que no forma una hélice sino una cadena en forma de zigzag. Del AA 53 al 53 encontramos una estructura helicoidal.
Estructura Terciaria
La Hb es casi esférica (globular), con un diámetro de 55 A, las cuatro cadenas están empaquetadas conjuntamente en disposición tetraédrica (ver anexo 2b). Los grupos Hemo, están localizados en unas oquedades cercanas al exterior de la molécula, uno en cada subunidad. Los 4 lugares de unión del oxígeno están separados, la distancia entre los dos átomos de Fe más próximos es de 25 A e inclinados con ángulos diferentes.
Cada grupo hemo se encuentra enterrado parcialmente rodeado por grupos -R Hidrofóbicos. Este se halla unido a la cadena polipeptídica mediante un enlace coordinado del átomo de Fe con la HIS (histidina), mientras el otro enlace de coordinación del Fe se halla disponible para el transporte del oxigeno. Existe una gran cantidad de residuos hidrofílicos en la superficie de la cadena, pero el centro de la a - hélice es especialmente hidrofóbico. Como en todas las proteínas existe una naturaleza anfipática, la cual hace que existan diferentes regiones que presenten mayor o menor polaridad, esto depende de los tipos de residuos que compongan la región. También existen fuerzas Vander Walls, aquellos que poseen un componente electrostático que se presentan cuando dos regiones apolares se encuentran lo suficientemente cerca para que se forme la fuerza entre los dipolos instantáneos o débiles (determinados residuos) y entre ellos producen un campo eléctrico, igualmente, las interacciones electrostáticas o puentes salinos, que se presentan cuando algunos iones se encuentran cercanos a la proteína y modifican el campo eléctrico, son importantes ya que estos dan estabilidad a la forma de la Hb.
Por último en la estructura de la Hb no hay enlaces de tipo S-S, ya que los residuos Cys (cistina), no son comunes en la cadena a aunque en la b si hay; pero no es posible que se establezcan este tipo de enlaces entre las cadenas a y b de las globinas. Cada cadena a está en contacto con las cadenas b, sin embargo, existen pocas interacciones entre las dos cadenas a o entre las dos cadenas b entre sí.
Estructura Cuaternaria
La Estructura cuaternaria modula las actividades biológicas de las proteínas. Tanto las proteínas transportadoras (hemoglobina), como las enzimáticas (A,T, C-ASA) pierden buena acción específica al fraccionarla en subunidades. La proteína íntegra al realizar la catálisis propia, admite una regulación en su actividad es decir puede frenarse o acelerarse, en respuesta a metabolitos concretos que pueden ser el propio sustrato o distintos moduladores alostericos, las propiedades alostéricas de la Hb se producen por la interacción de las subunidades diferentes. La unidad funcional de la Hb es un tetrámero que consta de dos clases de cadenas polipeptídicas.
La hemoglobina está clasificada dentro del grupo de las proteínas conjugadas ya que además de tener o poseer aminoácidos contiene además una proporción significativa del grupo prostético hem pues cada cadena del tetrámero está asociada a uno de estos. En el caso de la estructura de Hb se presenta el tercer caso que es el de los monómeros de función análoga pero de estructura diferente de tal manera que no se pueden sustituir unos por otros sin ciertas restricciones.
La asociación de diferentes tipos de globinas origina las diferentes especies tetraméricas de la Hb siendo HbAa 2b 2, HbA2a 2d 2, HbFa 2g 2. Se conocen como hemoglobinas anormales como la Hbb 4 o la HbBartz que es la d 4 con cuatro monómeros idénticos pero son funcionalmente inferiores a las antes mencionadas. En este caso no son posibles estructuras intermedias con estructura impar de monómeros de cada clase, ejemplo (a 3b ) Cuando se produce la oxigenación de la desoxihemoglobina, no hay variación alguna de la estructura terciaria; pero cuando se une el O2 a los grupos hemo de esta, las subunidades a , b que permanecen rígidas, cambian ligeramente de posición, aproximándose entre sí lo que presenta un cambio de la estructura cuaternaria. Existen dos clases de regiones de contacto entre las cadenas a y b . Uno de los tipos de contacto es (a 1b 2) que es idéntico al (a 2b 1); el otro tipo es (a 1b 1) y (a 2b 2). La estructura cuaternaria de la desoxiHb se denomina forma T tensa o tirante; la oxiHb forma R relajado. El átomo de hierro arrastra con él la histidina proximal cuando se introduce en el plano de la porfirina. Este movimiento de la histidina F8 provoca una alteración de la estructura hélice F y en los acodamientos EF y FG. Estos cambios conformacionales se transmiten a las interfases de las subunidades ocasionando la ruptura de los enlaces salinos intercatenarios lo que provoca que la proteína cambie a la forma R.
Esta estructura cuaternaria le confiere propiedades adicionales extraordinarias (ausentes en la hemoglobina) que la adaptan a sus papeles biológicos únicos y permiten una regulación precisa de sus propiedades. Las fuerzas que mantienen unidas las cadenas peptídicas para dar una estructura cuaternaria suelen ser de tipo fisicoquímico (asociación hidrofóbica), y el ensamblaje de monómeros se realiza espontáneamente, ocurre así la ordenación cuaternaria adoptada, representa un mínimo de energía libre para la molécula.
En este trabajo se hablará específicamente de un tipo de hemoglobinopatia muy común denomina Drepanocitos, anemia de células falciformes o de células en hoz, algunos datos históricos dan a conocer aunque existen datos contradictorios sobre el hecho, en la primavera de 1945 tuvo lugar durante un viaje en tren en Estados Unidos una conversación entre dos científicos destacados, William Castle y Linus Pauling, en la cual especularon sobre un posible origen de las enfermedades más allá del nivel celular, es decir, molecular, citando como caso posible la drepanocitosis. Este hecho cambiaría el curso de la historia de la medicina y permitiría descubrir el origen molecular de un trastorno de salud.
Cuatro años después, Paulin (único ganador en dos oportunidades del Premio Nobel junto a Marie Curie) reportó que la enfermedad de células en forma de hoz (falciformes) -descrita por primera vez por Herrick en 1910- podía tener origen en la alteración de una sola molécula del compuesto que transporta el oxígeno a los tejidos, la hemoglobina (del griego haimatos, sangre).
DREPANOCITIS
La drepanocitosis o anemia de hematíes falciformes es un padecimiento hereditario, ampliamente distribuido en todo el mundo como gen autosómico codominante. Los sujetos heterocigotos (AS) se designan como portadores, o que tiene el rasgo drepanocítico. Los homocigotos (SS) sufren de anemia drepanocítica.
Distribución geográfica.
Es probable que el padecimiento se haya originado en África, en donde en algunas zonas hay una prevalencia de portadores que oscila entre 20 y 40%. En Estados Unidos, de 0.1 a 0.2% de la población de origen africano sufre anemia drepanocítica (SS) y 8% es portador (AS) de la anormalidad.
Entre los descendientes de los esclavos de raza negra traída al Continente Americano, durante la época de la colonización Española, la prevalencia de heterocigotos en algunas regiones es análoga a la de África Tropical. En el Caribe, Centroamérica, Venezuela y Brasil, las anormalidades frecuente y, en algunos lugares, es muy elevada, como ocurre en ciertas localidades de Panamá, en donde los heterocigotos ocupan hasta el 30% de la población.
En México, Lisker y colaboradores demostraron que en ciertas zonas de las costas de Golfo y del Pacifico la Hb S es frecuente y existen algunas poblaciones con alta prevalencia de portadores (fig 1.1).
Algunos investigadores han propuesto que la mutación de la Hb otorga resistencia, pero no inmunidad a uno de los parásitos que causa el paludismo, Plasmodium Falciparum. Cuando los eritrocitos parasitados adoptan la forma semilunar o drepanocítica, el parásito muere. Por otro lado, las áreas geográficas en donde prevalece el P. Falciparum coinciden con las zonas donde han encontrado portadores de la Hb S.
Fisiopatología.
La Hb S (B6[A3]Glu Val) es el resultado de la sustitución de la base timina por la adenina en el codón 6 del gen B de globina (GAG TC) con sustitución del glutámico (GLU) por Valina (Val) (fig. 1.2). La localización superficial del aminoácido (residuo) mutado y su diferente carga eléctrica, explica que la Hb S pueda distinguirse fácilmente de la Hb A normal por su menor movilidad electroforética. Como consecuencia de esta mutación, cuando la hemoglobina se desoxigena (deoxi-Hb) sufre un proceso espontáneo despolimerización por lo que adopta la estructura de un gel cristalino conocido como cuerpo tactoide. La estructura de este polímero, se ha deducido a partir de estudios realizados mediante difracción de rayos X y microscopía electrónica de transmisión. Cada polímero está formado por catorce tetrámeros de deoxi-Hb que se disponen formando haces longitudinales unidos entre sí (Fig. 1.3). Esta alteración configura una estructura cilíndrica insoluble y rígida que modifica drásticamente la forma del eritrocito, el cuál adopta una morfología que recuerda a una hoz, el proceso de polimerización (drepanocitosis) no es instantáneo, si no que va precedido de un periodo de latencia durante el cuál las moléculas de deoxi-Hb establecen contacto (formación de pequeños agregado o nucleación) para finalmente polimerizar deforma explosiva en haces o fibras de cuerpos tactoides insolubles que vuelven rígidos a los eritrocitos. Estos cambios impiden que la sangre circule normalmente por los tejidos y se produce un estancamiento (fig 1.4). Este proceso requiere de un conjunto de factores facilitadores entre los de destaca mucho el descenso de la presión parcial de oxígeno (PO2) cuando esto sucede hay formación de cuerpos tactoides provocando el estancamiento y haciendo que la PO2 desciende más aún originándose un círculo vicioso: los eritrocitos falciformes incrementan el estancamiento, deciente más la PO2 y la falciformación se acentúa (fig. 1.5)
Otros factores facilitadores de la drepanocitosis son la concentración de Hb S (aumento de la CCMH), la disminución de la temperatura, la fuerza iónica del medio (disminución del pH) y la interacción de la Hb S con otras hemoglobinas normales ( Hb A, Hb A2 o Hb F) o patológicas (Hb C, Hb D, Hb O-Arab, Hb-J, principalmente). La interacción de la Hb S con otras hemoglobinas explica por qué la mezcla de cantidades proporcionales de Hb S y Hb A reduce la intensidad de la polimerización al 50% o que ésta sea nula si en lugar de Hb A existe Hb F. Este efecto de la Hb F sobre la polimerización es de gran interés, ya que explica la disminución de la expresividad clínica de la hemoglobinopatía S cuando coexiste con otras hemoglobinopatías, cono B y &B talasemias o la persistencia hereditaria de la hemoglobina fetal (PHHF). Además permite investigar opciones terapéuticas basadas en la inducción de síntesis de Hb F durante la edad adulta. Aunque el fenómeno de la falciformidad es reversible, entre el 5 y el 50% de los eritrocitos falciformes no pueden recuperar su forma original, por lo que son inmediatamente eliminados de la circulación por el mononuclear fogacítico (SMF). La proporción entre drepanocitos reversibles (DR) y drepanocitos irreversibles (DI) varía de un paciente a otro, aunque, generalmente, siempre existe un predominio de DR que recupera su forma normal (disco bicóncavo) en presencia de oxígeno. Los (DI) no recuperan la forma normal ni en presencia de oxígeno a elevada concentración y se caracteriza por presentar un elevado descenso del VCM (<70 fl) y aumento de CCMH(<370g/L). La concentración de CCMH pone de manifiesto un grado extremo de deshidratación celular que se explica por las alteraciones de polimerización irreversible de la deoxi-Hb s ejerce sobre la membrana eritrocitaria. Una de ellas es la alteración de sus propiedades fisicoquímicas. Así la formación de cuerpos tactoides intraeritrocitarios se acompaña de la formación de sustancias oxidantes (Ion superóxido, peróxido de hidrógeno, y radicales libres) que alteran la estructura de la membrana (modificación de composición y distribución de fosfolípidos en la bicapa) y condicionan un aumento de la permeabilidad pasiva al potasio y un exceso de calcio intraeritrocitario por el efecto Gardos). Otras alteraciones de la membrana del drepanocito son una levada tendencia a adherirse al endotelio vascular y una mayor sensibilidad al efecto de los fagocitos. Esta mayor adherencia de los drepanocitos al endotelio vascular resulta facilitada por sustancia como la trombospondina, resultado de la activación plaquetaria y la fibronectina. La trombospondina es una agente de adhesión especialmente activo debido a su afinidad por el antígeno CD36 presente en la membrana de los reticulocitos presentes en la crisis de anemia falciforme. Igualmente sucede con la fibronectina gracias también al mayor contenido de los reticulocitos en receptores alfa4B1(VLA 4). Esta mayor adhesión de los drepanocitos y reticulocitos al endotelio vascular constituye uno de los principales factores desencadenantes de las crisis vasooclusivas, características de la drepanocitosis.
Igualmente, el estímulo de la actividad macrofágica favorece la eliminación de los eritrocitos sensibilizados por el SMF.
La hemólisis de la anemia falciforme es intravascular y extravascular. El carácter intravascular resulta de la lisies de drepanocitos por acción del complemento (mayor sensibilidad de complemento). Y la pérdida de deformabilidad por la falciformación (mayor fragilidad al cizallamiento de la circulación sanguínea). El carácter de vasooclusión tiene un origen multifactorial y en su aparición pueden intervenir factores tan diferentes como la polimerización de la deoxi-Hb s, la pérdida de deformabilidad, y aumento de viscosidad sanguínea, mayor adherencia al endotelio vascular y activación de la hemostasia, variaciones de la tonicidad vascular, efectos directos de los granulocitos o plaquetas y también la presencia de factores facilitadores del entorno ambiental. De todos ellos, no obstante el que parece más importante es la adherencia de los drepanocitos al endotelio vascular. Este proceso puede ser desencadenado por un proceso infeccioso o inflamatorio, con activación de granulocitos y plaquetas. Ambas activación es por mecanismos diferentes, pero convergentes, aumentan la adhesión de los eritrocitos al endotelio vascular y junto con la disminución de la deformabilidad, facilitan la obstrucción vascular y con ello la crisis vasooclusiva. Hay que señalar que la vasooclusión constituye un factor local que implica por efecto de la hipoxia local el proceso de la falciformación y enlentecimiento de la circulación sanguínea local.
Manifestaciones clínicas
Existen dos formas clínicas de Hb S: Homocigoto (HbSS), en la que los pacientes sufren anemia falciforme (anemia hemolítica y crisis vasooclusivas), y heterocigótica (HbAs), generalmente asintomática. La forma más frecuente de la Hb S es el rasgo heterocigoto, del que se cree que existe en el mundo mas de 35 millones de individuos afectados. La intensidad de las manifestaciones clínicas puede depender también de la coexistencia de otras hemoglobinopatías asociadas.
Hemoglobina (S) o forma heterocigota (portadores o rasgo drepanocítico)
Los sujetos se hallan habitualmente asintomáticos y la exploración física es negativa. No obstante, es conveniente descubrirlos porque algunos pueden llegar a sufrir hematurias secundarias a necrosis papilar renal; otros padecen infartos de bazo o trombosis cerebrales. Estas complicaciones casi siempre ocurren cuando los portadores se exponen a situaciones de hipoxia prolongada, como en las anestesias generales con inadecuada aportación de oxigeno, en los procesos neumónicos y durante los viajes en aviones desprovistos de cabina con presión. La exposición al frío intenso puede precipitar también estas complicaciones. Como estas últimas son excepcionales, se ha recomendado considerar a los portadores como personas sanas, pues se ha comprobado que muchos de ellos pueden tolerar esfuerzos físicos intensos. En apoyo a esto, cabe señalar que cerca del 7% de los jugadores de raza negra de la Liga Nacional de Fútbol Americano de Estados Unidos son portadores de Hb S. Se conoce también el vaso de un excampeón de boxeo, portador de la anormalidad.
Datos de laboratorio. Casi nunca existe y la morfología de los eritrocitos es normal. De manera excepcional se encuentran drepanocitos en los extendidos convencionales se sangre. Para descubrir a los portadores se requiere practicar pruebas de inducción de los drepanocitos y de solubilidad que son positivas. La electroforésis de hemoglobina en acetato de celulosa, pH 8.4-8.6, que nunca debe omitirse, puede demostrar el componente S de la hemoglobina, de motilidad electroforética más lenta que la Hb A. La combinación de un patrón electroforético AS y una prueba de solubilidad positiva establecen el diagnóstico de portador (AS) de esta variedad de hemoglobina.
Hemoglobina (S) homocigota (SS) o anemia drepanocítica.
En el caso de Hb SS la intensidad del cuadro clínico varía ampliamente de un paciente a otro y su inicio debido al efecto protector de la Hb F durante el periodo neonatal, es siempre pasado de 6 a 4 meses de vida.
Los enfermos, en su mayoría de raza negra con rasgos negroides, generalmente son altos, delgados, con cráneo en torre y articulaciones hiperextensibles (fig 1.6). Su crecimiento y maduración sexual están generalmente retardados. Al examinarlos se aprecia ictericia conjuntival y no es raro que este dato desoriente al médico no familiarizado con esta enfermedad y establezca el diagnóstico clínico de hepatitis. Los enfermos sufren úlceras maleolares o cicatrices de úlceras antiguas. En el fondo de ojo, los vasos retinianos se encuentran tortuosos o en “tirabuzón”.
Las crisis vasculares oclusivas son características de la afección, resultado de la oclusión de los vasos por acumulación de drepanocitos que origina estancamiento e infarto.; aquellas ocurren a cualquier edad y tienen, como denominador común, antecedentes de hipooxigenación excesiva, como la secundaria a procesos infecciosos, deshidratación, ejercicios violentos, trabajo de parto y grandes altitudes, como a las que se exponen las personas cuando viajan en aviones desprovistos de cabina de presión. La fiebre moderada menudo se asocia con crisis vasoclusivas.
En los niños de nueve meses de edad, las crisis oclusivas que afectan a los huesos pequeños de manos y pies determinan dactilitis o síndrome de “mano-pie” (fig. 1.7), caracterizado por la inflamación dolorosa del dorso de estas regiones. A menudo ésta es la primera manifestación clínica de la enfermedad.
Las crisis aplásticas son resultado de la interrupción brusca de la eritropoyesis, con descenso de la reticulocitosis y de los niveles de hemoglobina. La caída de ésta última puede ser muy grave y causar insuficiencia cardiaca y muerte en pocas horas. Las crisis aplásticas se han asociado a diversas causas. Sin embargo, algunos estudios epidemiológicos han demostrado infecciones por parvovirus B19 constituyen la causa de la mayoría de ellas, y es posiblemente la de todas, las crisis aplásticas resultantes de la toxicidad directa del parvovirus en los precursores eritroides, especialmente de las unidades formadoras de colonias eritrocíticas.
En el desarrollo de de la anemia falciforme pueden considerarse tres fases evolutivas con sintomatologías características: a) Fase estacionaria. B) Fase de expresividad aguda y fase de expresividad crónica.
Fase estacionaria.
Corresponde generalmente a los primeros años de vida (uno a cuatro años) y sus manifestaciones clínicas son las propias de un síndrome hemolítico crónico moderado o intenso, (anemia, palidez cutaneomucosa, subictericia conjuntival y retraso del crecimiento óseo y gonadal). En esta fase es característica una intensa retención eritrocitaria esplénica (hiperesplenismo) con complicaciones vasooclusivas de carácter local y progresivo que condicen a la pérdida de la función esplénica o autoesplenectomía. Esta puede ponerse fácilmente de manifiesto mediante la visualización de vacuolas (pits) eritrocitarias cuando se observa una suspensión de sangre con glutaraldehido mediante microscopia convencional y óptica de NOMARKI
Fase de expresividad aguda
Se inicia a partir de los cuatro años de edad con agravamiento agudo del cuadro anémico (Hb <80g/L) y aparición de diversas manifestaciones clínicas de carácter agudo debidas a la crisis vasooclusivas que afectan de forma importante diversos órganos aunque muy especialmente pulmón, riñón, y tejido óseo. Estas se caracterizan por un dolor muy intenso en los territorios afectados (tórax, huesos, y zonas distales de las extremidades), generalmente acompañadas de infecciones que suelen ser recidivantes.
Las crisis de dolor agudo constituyen, de hecho, la manifestación más característica de la anemia falciforme y muchas veces su primer síntoma. Aunque pueden aparecer espontáneamente, suelen ser desencadenadas por situaciones tan diversas como hipoxia, fiebre, infecciones, deshidratación, frío., cambios estacionales o menstruación. Obedecen a oclusiones de la microvasculatura que pueden afectar distintos territorios del organismo (crisis vasooclusivas). Una de sus manifestaciones más características es el dolor óseo generalizado o limitado a huesos largos (húmero, fémur, tibias) en sujetos adultos o pequeños de las extremidades superiores e inferiores (dactilitis) en los niños. La dactilitis da lugar al conocido síndrome mano-pie, consiste en un dolor agudo y muy intenso con tumefacción subcutánea de la superficie dorsal de las manos y pies acompañado de impotencia funcional. Este síndrome puede confundirse fácilmente de con acceso de fiebre reumática o artritis séptica.
Otras regiones que también suelen afectarse en las crisis dolorosas son la condrocostal (dolor torácico), vertebral (dolor dorsolumbar) y el vaso (cuadro de dolor abdominal agudo). En un gran número de pacientes, la tomografía computarizada (TC) y la gammagrafía ósea permite detectar precozmente estas lesiones. Otro territorio también afectado por las crisis vasooclusivas es el mesenterio, dando lugar a infartos de vasos mesentéricos que son causa de dolor abdominal muy intenso y de carácter agudo (síndrome mesentérico). Igualmente, la oclusión de los vasos cerebrales es causa frecuente de accidente neurológico agudo como hemiplejía, monoplejía y convulsiones.
Las infecciones constituyen la complicación de la anemia falciforme y las responsables de un elevado porcentaje de fallecimientos en estos pacientes. Ello obedece a que solo constituyen una complicación del cuadro drépanocitico sino que son, en sí mismas, un factor desencadenante de la crisis. La incidencia de las infecciones va disminuyendo progresivamente con la edad, ya que en los niños, está facilitada por la pérdida de la función inmunitaria del bazo como consecuencia de la autoesplenectomía por micro infartos de repetición (hiposplenia o asplenia), aunque en los menores de cinco años, la hepatoesplenomegalia es frecuente pero, a medida que se producen infartos en el bazo, se reduce su tamaño y la víscera acaba por desaparecer. Esto es tan frecuente que cuando se encuentra esplenomegalia en los enfermos de más edad, hay que dudar si verdaderamente seas homocigotos (SS) o dobles heterocigotos (heterocigotos compuestos), como es el caso de pacientes con las combinaciones genéticas SC, SD y S-talasemia B, en quieres la autoesplenectomía no ocurre.
Como infecciones más frecuentes destacan las debidas a S.Pneumonidae y H.Influensae, aunque también suele observarse osteomielitis producida casi siempre por bacterias del género salmonella. La complicación infecciosa más grave del cuadro drepanocítico es la septicemia posmeningitis por S.Pneumonidae o H.Influensae, cuyas manifestaciones clínicas, siempre muy aparentes, suelen asociarse a coagulación intravascular diseminada (CID). Con menos frecuencia pueden aparecer infecciones pulmonares de carácter muy grave con complicaciones tromboembólicas casi siempre producidas por M.Pneumonidae.
La aparición de fiebre taquipnea e incluso dolor torácico (síndrome torácico) constituye la causa más frecuente de hospitalización con anemia falciforme. El síndrome torácico pone de manifiesto una afección del sistema vascular pulmonar que casi siempre se acompaña de infección o hipertensión pulmonar y que suele cursar con una insuficiencia cardiorrespiratoria que puede causar la muerte.
Menos frecuentes que las complicaciones pulmonares son las oclusiones vasculares agudas de otros territorios en los que destacan la retina, el sistema nervioso central (SNC) y los cuerpos cavernosos del pene. La trombosis de la arteria central de la retina puede constituir una causa de ceguera (amaurosis) y la de los cuerpos cavernosos del pene de priapismo que, con el tiempo, se transforma en impotencia por fibrosis de los tejidos esponjosos eréctiles del pene. Ambas complicaciones son en general menos frecuentes que la trombosis cerebral, causa relativamente frecuente de apoplejía.
Fase de expresividad crónica
Es propia de los pacientes que han logrado sobrevivir la primera infancia, por lo que es característica de la adolescencia y edad adulta (tabla).
El carácter evolutivo crónico de la anemia falciforme afecta de forma importante el crecimiento y desarrollo corporal, el sistema nervioso central, cardiovascular, pulmonar, hepatobiliar y gastrointestinal. Asimismo, condiciona lesiones de función renal y trastornos visuales que pueden conducir a la ceguera. Finalmente, otra complicación relativamente frecuente de la drepanocitosis en su fase crónica son las úlceras maleolares de evolución tórpida.
Retraso del crecimiento y lesiones osteoarticulares
Una de las manifestaciones más evidentes de la drepanocitosis en su fase crónica es el retraso de crecimiento, puesto de manifiesto por un peso y altura inferiores a los que corresponden a la edad cronológica. Ello se acompaña de un retraso de desarrollo gonadal con hipogonadismo y aparición tardía de la pubertad y la menstruación. También la cronificación de los episodios agudos de dolor conduce a una destrucción progresiva de los huesos y articulaciones afectadas con aparición de osteonecrosis en epífisis de huesos largos (cabeza del fémur) y vértebras (aplastamiento vertebral) junto a derrames articulares que pueden acompañarse de dolor intenso, fiebre y leucocitosis. Con menor frecuencia puede observarse también afectación de la calota craneal con engrosamiento del diploe e imagen radiológica en cepillo, cuyas características son superponibles a las que se observan en la talasemia mayor.
Todas estas alteraciones óseas pueden ponerse fácilmente de manifiesto mediante examen radiológico o gammagrafía ósea.
Sistema nervioso central
Durante la fase crónica, aunque pueden observarse crisis de apoplejía, los trastornos más comunes son la existencia de cierto retraso psicomotor que explica las dificultades de aprendizaje y el déficit neuropsicológico que suelen presentar estos pacientes durante la edad escolar. Para evitar lo posible estas complicaciones, resultado del sufrimiento cerebral crónico, es muy recomendable prevenir en lo posible su desarrollo mediante técnicas que determinan la fluidez de la circulación cerebral, por ejemplo el Doppler transcraneal o que facilita su observación precoz, como la tomografía computarizada (TC) o la resonancia magnética (RM)
Aparato cardiocirculatorio
La cronicidad de la anemia falciforme suele acompañarse de una cardiomegalia e insuficiencia ventricular izquierda, en cuyo desarrollo pueden intervenir varios factores, como los infartos múltiples de arterias pulmonares y miocárdicas, la hemosiderosis miocárdica postransfusional y la hipertensión arterial secundaria a la insuficiencia renal. En estos casos, la ecocardiografía pone de manifiesto el aumento de tamaño ventricular y la sobrecarga funcional cardiaca impuesta por la anemia crónica y la hipertensión arterial. En ocasiones, aparecen signos clínicos de infarto agudo del miocardio sin afectación coronaria o aterosclerosis.
Sistema pulmonar
En la fase crónica de la anemia falciforme es frecuente observar signos de insuficiencia respiratoria obstructiva secundaria a la afección pulmonar por micro infartos de repetición y sobreinfecciones que desembocan con el tiempo en una fibrosis pulmonar progresiva. En este trastorno al favorecer la hipoxia, contribuye a incrementar la frecuencia de las crisis de drepanocitosis y con ello agravar el cuadro clínico.
Sistema hepatobiliar
En la anemia falciforme la hepatomegalia constituye un signo clínico prácticamente constante, casi siempre consecuencia del proceso hemolítico crónico (hemosiderosis) y de infecciones víricas postransfusionales. Esta hepatopatía se acompaña de signos biológicos de afección hepatobiliar (aumento moderado de las transaminasas o fosfatasa alcalina del plasma) e histológicos de eritrofagocitosis con aumento de pigmentos de hemosiderina y una grado variable de fibrosis periportal, otra consecuencia hepatobiliar de la hemólisis crónica e hipercatabolismo de la hemoglobina es la litiasis biliar, que afecta a un 10-15% de los pacientes.
Función Renal
Las complicaciones renales de la anemia falciforme son relativamente frecuentes y obedecen mayoritariamente a la necrosis papilar que aparece como consecuencia de microtrombosis en la región de las asas de Henle. El aumento local del hematocrito, osmolaridad y la disminución del Th y PO2 hacen de esta región un lugar idóneo para la falciformación y microinfartación de las papilas y pirámides de la pelvis renal. La consecuencia fisiopatológica de este trastorno es la pérdida de la capacidad renal para concentrar y diluir la orina (hipostenuria9 y la eventual aparición de hematuria macroscópica. Esta última puede ser también consecuencia de una glumerulonefritis estreptocócica, complicación relativamente frecuente de la Hb S14. El síndrome nefrótico, aunque posible, no constituye una complicación tan frecuente como la anterior y suele acompañarse de hipertensión , hematuria y disminución progresiva de la función renal. Incluso en ausencia de manifestaciones clínicas y afectación renal, un elevado número de pacientes con anemia falciforme presentan proteinuria y un moderado aumento de la creatinina plasmática lo que pone de manifiesto la frecuencia de la afectación renal crónica en ésta enfermedad. Aunque la insuficiencia renal aparece solo en un 4% de pacientes con anemia falciforme, constituye una importante causa de muerte en la edad adulta. La hemodiálisis y la administración de eritropoyetina recombinante (HrEpo) en caso de anemia intensa, constituyen, en este caso, medidas terapéuticas de elección.
Trastornos oculares
Las complicaciones oculares obedecen a oclusiones de los pequeños vasos de la retina que ocacionan subfusiones hemorrágicas y signos proliferativos característicos de neoformación vascular compensadora. Debido a ello se produce una retinopatía proliferativa cuyas características y evolución son muy similares a las de la retinopatía diabética: alteraciones de la visión con la aparición de fotopsias, moscas volantes, y adherencias corioretinianas que con el tiempo pueden facilitar el desprendimiento de la retina. Otra complicación relativamente frecuente, de la anemia falciforme, es el hifema o su fusión hemorrágica en la cámara anterior de globo ocular.
Ulceras maleolares
Este trastorno se ha descrito siempre como una compli8caciñon característica de la anemia falciforme, ya que se observa en más del 50% de pacientes. Constituye una complicación menos grave en comparación con las mencionadas anteriormente y su aparición resulta favorecida por traumatismos y ciertas infecciones (linfangitis y osteomielitis) aunque no son muy dolorosas las úlceras presentan un carácter torpido que dificultan su resolución definitiva.
Impotencia
Consecuencia de la fibrosos de los cuerpos cavernosos del pene como resultados de las trombosis crónicas con priapismo.
Rasgo falciforme
El rasgo falciforme corresponde al fenotipo de los portadores heterocigotos del gen B5 y se caracterizan por la ausencia de signos clínicos de la enfermedad. La hemoglobinopatía solo puede ponerse de manifiesto mediante procedimientos de laboratorio (electroforesis de Hb). Por ello, cuando en un individuo portador de rasgo falciforme se aprecian signos clínicos de hemólisis es prudente revisar de nuevo el diagnóstico por si tal manifestación pudiera corresponder a otra causa, el examen morfológico de los eritrocitos no muestra alteración alguna y la concentración en sangre de reticulocitos, así como los índices eritrocitarios (BCM, HCM y CCMH), son normales. Excepcionalmente la enfermedad puede manifestarse en situaciones de estrés como disminuciones importantes de la PO2, altitud, submarinismo o anestesia., o tras la realización de un ejercicio físico intenso ésa última posibilidad podría explicar la muerte súbita de deportistas o soldados después de una marcha extenuante en cualquier caso, las manifestaciones clínicas del rasgo heterocigoto cuando existen se refieren más a alteraciones vasooclusivas que a la anemia hemolítica. Así, una de ellas es la afectación del riñones (hipostenuria con hematuria) por microinfarto de la médula renal o el dolor abdominal agudo por microinfarto esplénico.
Diagnóstico de laboratorio
La confirmación diagnóstica de la anemia falciforme o de su carácter portador. Precisa la realización de diversas pruebas de laboratorio en las que se destacan: el hemograma, la electroforesis de hemoglobinas a pH alcalino y las pruebas de solubilidad de Hb y de la falciformación.
El hemograma muestra una anemia normocítica, o ligeramente macrocítica, con valores de Hb que suelen oscilar entre 70 y 90 g/L y una reticulocitosis casi siempre superior a 150 x 10-9/L y que, en ocasiones, puede llegar a 600 x 10-9/L. en individuos adultos con Hb SS, el VCM y el CCMH mediante sistemas independientes. En cualquier caso en examen morfológico de la extensión de sangre muestra una proporción variable de eritrocitos falciformes, junto a una leucocitosis neutrofílica moderada (15-30x109/L) y ligera trombocitosis.
La prueba de solubilidad es el procedimiento más simple para complementar la información aportada por el hemograma y el examen morfológico de los eritrocitos, esta prueba consiste en observar la precipitación de la Hb S cuando el bemolizado se incuba en presencia de bitionito sódico que actúa como agente reductor. La prueba de la solubilidad puede complementarse con la llamada prueba de desoxigenación que consiste en inducir la formación de eritrocitos falciformes in Vitro, exponiendo la sangre total a un estado de hipoxia. Para ello se coloca una suspensión de eritrocitos sobre un portaobjetos, se añade una agente desoxigenante (metabisulfito o ditionito sódico al 2%) y se cubre con un cubreobjetos sellando las esquinas para evitar en lo posible el contacto de la sangre con el O2 ambiental, pasados unos minutos se observan en un microscopio la posible inducción de eritrocitos falciformes. La erectroforésis de Hb a Ph alcalino es el procedimiento diagnóstico que por su simplicidad, precisión y bajo costo ofrece un mayor rendimiento. Empleando esta técnica los individuos homocigotos muestran una fracción de Hb mayoritaria que migra algo por detrás de la Hb F y ausencia de Hb A. en caso de portadores heterocigotos se aprecian dos fracciones hemoglobínicas de intensidad simi8lar y un moderado aumento de HbF en ambas situaciones la Hb A2 es normal. El empleo de isofocalización y cromatografía líquida de alta resolución (CLAR) puede ser útil para la realización de estudios familiares extensos o escrutinio de loa hemoglobinopatía.
Los problemas diagnósticos en casos de anemia falciforme surgen cuando junto a la Hb S coexisten otras hemoglobinopatías estructurales o talasemias. La asociación de hB sC no suele crear problemas ya que le electroforesis permite apreciar claramente las dos fracciones, S y C muy bien separadas. En caso de Hbs-talasemia, por el contrario pueden existir dificultades. Estas aparecen caso de Hb S SB-talasemia, donde existe un claro predominio de la Hb F sobre la A normal, (10-30%). Esta situación se diferencia de la drepanocitosis heterocigoto, caracterizada por un moderado predominio de la Hb A sobre la S, en caso de la Hb SB-0-talasemia y Hb S A-Talasemia el patrón electroforético es idéntico al de la drepanocitosis homocigoto, excepto que en ambos casos suele observarse un aumento de la Hb A-2, junto a la disminución del BCM. Por ello en ambas situaciones el diagnóstico diferencial exige el empleo de la biología molécular aunque ninguna de ella debe olvidarse de una transfusión reciente puede falsear el patrón electroforético y hacerse muy similar al de la drepanocitosis heterocigoto, debido a la perfusión de eritrocitos normales que solo contienen HbA.
En resumen
El diagnóstico de laboratorio de hemoglobinopatía S se establece con los siguientes estudios:
Prueba de inducción de los drepanocitos positiva o, mejor aún.
Prueba de solubilidad positiva. Hay que recordar que todos los enfermos con genotipo S (SS, AS, SC, SD y S-talasemia B) tienen esta prueba positiva.
El estudio electroforético de la hemoglobina en acetato de celulosa, pH 8.4-8.6 compatible con el genotipo SS.
Determinación de Hb F generalmente aumentada (10-20%)
Fracción A2 de hemoglobina dentro de los límites normales.
Prevención y pronóstico de la anemia falciforme
La incorporación del diagnóstico precoz la educación sanitaria, y las posibilidades terapéuticas, has significado una sensible mejora en el pronóstico y calidad de vida en los pacientes afectados de drepanocitos homocigoto, es un hecho bien conocido que este trastorno presenta características clínicas muy variable, en unas áreas geográficas es prácticamente asintomático en otros reviste la gravedad de las crisis vasooclusivas y sus complicaciones. Actualmente se conocen los tres moduladores de la expresión clínica sobre los que se podría intentar actuar con finalidades terapéuticas : concentración de hb F, coexistencia de genes talasemicos y el sexo. La Hb F no participa en el proceso de polimerización de la desoxi-Hb S, por lo que cuando su concentración intraeritrocitaria es elevada existe una menos tendencia ala falciformación, resulta evidente, por tanto, que la concentración de Hb F constituye un factor de gran importancia pronostica de la anemia falciforme. La mayor o menos concentración de Hb F comprende de varios factores en gran parte constitucionales entre ellos destacan la edad, sexo, el numero de genes A, los aplotipos del gen B, y la actividad del locus FCP. El locus FCP es un gen que regula la producción de Hb F en los pacientes con anemia falciforme y se haya situado en el cromosoma sexual X (Xp22.2) lo que explica que la producción de Hb F sea superior en mujeres independientemente de que sean portadores del gen Bs. Debido a ello, las mujeres con anemia falciforme portadoras del aplotipo poseen mayor Hb F que los varones con idéntico aplotipo la existencia de polimorfismos en el gen beta de globina parece determinar las diferencias de comportamiento clínico entre razas africanas, mediterráneas y asiáticas, así, mientras en las proporciones africanas y del mediterráneo el aplotipo predominante es el Benin, en las asiáticas es el cenagal. La asociación de este ultimo a una mayor síntesis de Hb F explica su mejor comportamiento clínico y menor predisposición a padecer crisis vasooclusivas o sus complicaciones la coexistencia en un mismo individuo de un gen beta 5 alfa talasemia, es poco frecuente y se caracteriza pro una menor anemia y un moderado aumento de la fracción Hb A2 con valores de VCM y CCMH algo disminuidos. Este comportamiento hematológico junto con una menor proporción de drepanocitos irreversibles, explica la relativa benignidad del cuadro clínico y su mejor pronóstico, por el contrario, la coexistencia de alfa-talasemia, al acompañarse con un valor de hematocrito más elevado puede tener un riesgo mayor de crisis vasooclusivas, en cualquier caso una de las mejores opciones para estos pacientes es prevenir en lo posible la aparición de cuadros agudos de vasooclusión ya que constituyen la manifestación clínica más grave de dicha anemia. Para ello el Sickle Cell Information Center de Georgia (EEUU) propone seguir las pautas que brevemente se prescriben a continuación:
Tomar ácido fólico diariamente para garantizar el funcionamiento de la eritropoyesis
Administración de penicilina con carácter preventivo hasta los 6 años de edad para evitar en lo posible las infecciones graves.
Mantener un buen estado de hidratación mediante la ingestión de 8 a 10 vasos al día en adultos.
Evitar en lo posible ambientes excesivamente fríos o calurosos.
Evitar ejercicio intenso y estados de estrés.
Procurar descansar al máximo
Realizar controles médicos periódicos
Tratamiento
La anemia falciforme debe ser tratada siempre teniendo en cuenta su carácter crónico, y la frecuencia de sus complicaciones, por ello, el tratamiento debe ser ante todo preventivo, sobre todo con el consejo genético y explicar los puntos importantes sobre su transmisión familiar:
También prevenir evitando las situaciones que favorecen loas crisis vasooclusivas como son las infecciones, acidosis, hipoxemia y la exposición al frío, si no puede evitarse las crisis agudas, con aparición de dolor y fiebres altas lo más pudente es la hospitalización del paciente para mejorar su estado de oxigenación, evitar la deshidratación y administrar antibióticos. Cuando se presente deshidratación o como prevención se administrará por vía parenteral o digestiva, liquido en cantidad de 2 a 4 L/m2 cada 24H.
Infecciones
La elevada frecuencia de infecciones neumococicas en la anemia falciforme especialmente en niños de corta edad obliga a la inmunización, los principales agentes, se dará administración penicilina VP cada tres semanas, el peligro de sobreinfecciñón vírica transfucional puede prevenirse mediante de epavax. Junto a ello es aconsejable la administración de una dosis de penicilina
Anemia
Los pacientes con anemia falciforme presentan un elevado grado de adaptación a ella inclusive para valores muy bajos de concentración de Hb, en ocasiones no obstante la evolución en crisis de anemia agua hacen necesarios recurrir a las transfusiones de sangre total o de eritrocitos esta practica debe indicarse en los casos solo imprescindibles, ya que como es conocido, las transfusiones son causa de complicaciones como inmunización alogénica, hemosideroris e infecciones víricas. En general consisten en la administración de concentrados eritrocitarios hasta conseguir valores de concentración de Hb en sangre entre 100-120/L. es muy importante no superar este limite ya que podrirán aparecer complicaciones por aumento de viscosidad y bolemia en la sangre, se requiere administración simultánea de diuréticos.
Agentes antidrepanociticos.
Desde hace tiempo se han ensayado la administración por vía oral y parenteral de compuestos de triple finalidad de disminuir la polimerización de desoí-Hb S, aumentar la afinidad de la hemoglobina por el oxígeno y mejorar las propiedades biológicas eritrocitarias. Estos compuestos, conocidos como agentes antidrepanociticos, han basado su acción en un aumento de la síntesis de Hemoglobina fetal o en una inhibición de la polimerización de desoí-Hb s a través
Datos de laboratorio. La anemia, habitualmente normocítica normocrómica, es grave. Oscila entre 5 a 9g/dL, se inicia entre los seis y nueve meses de edad y se mantiene toda la vida. La anisocitosis es frecuente y se observan numerosos dianocitos (eritrocitos en forma de “diana”)y drepanocitos. Los ovalocitos y esquistocitos (eritrocitos fragmentados) también se encuentran. Se observan, asimismo, policromatofilia, eritrocitos nucleados (normoblastos) y reticulocitosis. Como en todas las anemias hemolíticas, se demuestra hiperbilirrubinemia indirecta (no conjugada); incremento de la deshidrogenasa láctica; disminución de los niveles de haptoglobina en suero; acortamiento de la sobrevida de los eritrocitos, e hiperplasia eritroide en médula ósea.
Tratamiento.
Por ahora no se dispone de ningún tratamiento específico. Ciertas medidas profilácticas y precauciones generales contribuyen a reducir las crisis, como evitar cambios bruscos de temperatura, la deshidratación, y las infecciones. Entre estas últimas las ocasionadas por Streptococcus pneumoniae pueden evitarse con el uso de una vacuna específica y la aplicación profiláctica de penicilina de acción retardada, particularmente en niños de cuatro meses a cinco años. Las vacunas en contra del Haemophilus influenzae, virus de la influenza y de hepatitis b, ofrecen protección adicional a los pacientes. Como en todos los procesos hemolíticos crónicos, debe administrarse ácido fólico permanentemente para impedir su déficit. Dentro de estas medidas profilácticas puede incluirse la detección intrauterina de homocigotos, empleando técnicas de ADN recombinante, cada día más perfeccionadas y con menor riesgo para el producto, pero aún limitadas a los centros que las practican.
Las crisis vasoclusivas se tratan con reposo, rehidratación, oxigenación y analgésicos. Las infecciones se investigan por medio de cultivos pertinentes y se tratan con antibióticos y agentes quimioterápicos requeridos. La administración de eritrocitos empacados se utiliza cuando los síntomas y signos físicos lo sugieran. Hay que tener en cuenta que muchos pacientes que padecen anemia crónica toleran muy bien las cifras muy bajas de hemoglobina.
De algunos años a la fecha ha sido estudiado el empleo de varios compuestos químicos que impiden la polimerización de la hemoglobina S o preservan la integridad de la membrana de los eritrocitos. Desafortunadamente no se ha encontrado aún el medicamento idóneo, pero todo hace suponer que a la postre habrá de descubrirse.
Entre los fármacos empleados pueden señalarse intravenosa, el cinato de sodio, el nitrógeno de mostaza y los compuestos que preservan la integridad de la membrana como zinc, el cetiedil, el telurito. El transplante de médula ósea constituye una alternativa para algunos pacientes. El consejo genético debe proporcionarse en parejas de riesgo.
6. Bibliografía
http://www.araucaria2000.cl/scirculatorio/sistemacirculatorio.htm
http://www.saludhoy.com/htm/adoles/articulo/drepanoc.html
BOHINSKI, Robert. Bioquímica. Edit. Fondo Educativo Interamericano. 1976
LA BASE MOLECULAR DE LA VIDA. Blume Ediciones. 1978
MACARULLA, Jose. Biomoléculas: Lecciones de Bioquímica estructural. Editorial Reverté.1978 España
STRYER, Lubert. Bioquímica, Tomo 1. Edit. Reverté, 4º edición
1
Fig., 1.2 Hemoglobina S
Fig. 1.3 Cuerpos tactoides
Con microscopía electrónica los polímeros de HbS se ven como haces de 14 fibras que se orientan en el sentido longitudinal de la célula.
La velocidad y extensión del polímero depende de tres variables independientes:
1.-Del grado de oxigenación de la célula.
2.-De la concentración intracelular de Hb.
3.-De la presencia o ausencia de HbF.
Fig. 1.7 Síndrome de “mano-pie” en un paciente con anemia drepanocítica.
Descargar
| Enviado por: | Berem |
| Idioma: | castellano |
| País: | México |
Todos los derechos reservados.